Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Here is a thorough synthesis:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of non-cardiogenic pulmonary edema driven by diffuse lung inflammation, increased alveolar-capillary permeability, and respiratory failure. Unlike hydrostatic (cardiogenic) edema, the edema fluid in ARDS is protein-rich and exudative, reflecting barrier breakdown rather than elevated hydrostatic pressure.
1. Inciting Triggers
ARDS arises from two broad categories of insult:
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia, aspiration of gastric contents | Sepsis (most common) |
| Pulmonary contusion, toxic inhalation | Severe trauma, multiple fractures |
| Near-drowning | Massive transfusions (TRALI) |
| — | Pancreatitis, drug overdose, burns |
The Berlin definition requires: bilateral opacities on chest imaging, PaO₂/FiO₂ ratio <300 mmHg (on ≥5 cmH₂O PEEP), onset within 7 days of a clinical insult, and pulmonary edema not fully explained by cardiac failure or fluid overload. — Murray & Nadel's Textbook of Respiratory Medicine
2. Three Sequential Pathological Phases
Phase 1 — Exudative Phase (Days 1–7)
The hallmark pathology is diffuse alveolar damage (DAD):
- Injury to alveolar capillary endothelial cells and type I pneumocytes breaks down the normally tight alveolar-capillary barrier
- Protein-rich edema floods the interstitium and alveolar spaces
- Condensed plasma proteins aggregate with cellular debris and dysfunctional surfactant to form hyaline membrane whorls lining the denuded alveolar walls
- Widespread neutrophil infiltration of the interstitium and airspaces occurs
- Pulmonary vascular injury leads to microthrombi and vascular obliteration
(Note: DAD is confirmed in only ~50% of ARDS patients on autopsy; the remainder may have pneumonia, diffuse alveolar hemorrhage, or eosinophilic pneumonia.)
Phase 2 — Proliferative Phase (Days 7–21)
- Hyaline membranes are reorganized
- Type II pneumocytes proliferate in an attempt to restore the epithelial barrier
- Fibrosis begins to appear; obliteration of pulmonary capillaries and deposition of interstitial/alveolar collagen are observed
- Neutrophil numbers decrease and pulmonary edema begins to resolve
Phase 3 — Fibrotic Phase (>2–3 weeks)
- A subset of patients develop pulmonary fibrosis
- Notably, fibroproliferation may begin simultaneously with early inflammatory injury — elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid within 24 hours of ARDS onset
(CT correlates of all three phases are shown below)
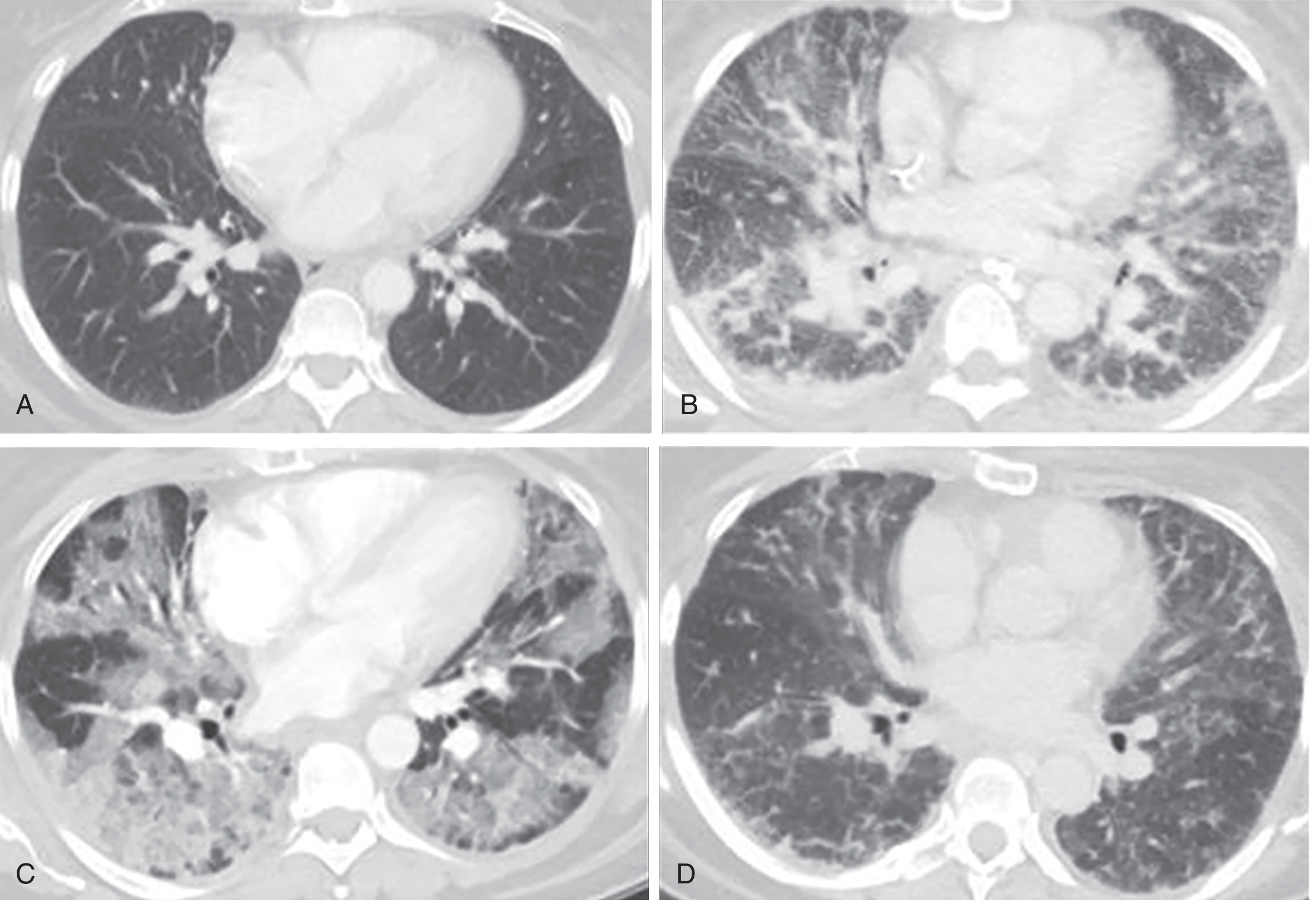
3. Cellular and Molecular Mechanisms
3a. Alveolar-Capillary Barrier Breakdown
The alveolar-capillary membrane consists of:
- Microvascular endothelium — loss of endothelial barrier integrity is both necessary and sufficient for ARDS development
- Alveolar epithelium — damage to type I pneumocytes (which cover ~95% of the alveolar surface) is the key precipitating event
Mechanisms of epithelial and endothelial cell death include necrosis, apoptosis, mechanical stretch-induced injury, and coagulation-related death. Loss of epithelium has a double effect: it both (1) disrupts the barrier, promoting edema, and (2) prevents active alveolar fluid clearance. — Murray & Nadel's Textbook of Respiratory Medicine
3b. Neutrophil-Mediated Injury
Neutrophils are central effectors:
- Activated neutrophils are sequestered within the alveolar and interstitial spaces in massive numbers
- They degranulate, releasing:
- Proteases (elastase, matrix metalloproteinases) — degrade the extracellular matrix and tight junctions
- Reactive oxygen species (ROS) — cause oxidative damage to cell membranes
- Myeloperoxidase (MPO) — amplifies oxidative injury
- Neutrophil extracellular traps (NETs) — directly injure endothelial and epithelial cells
- Neutrophil elastase also degrades surfactant protein A, impairing host defense and surface tension regulation
- Platelet-neutrophil complexes (formed via P-selectin / PSGL-1 binding) amplify this injury cascade, triggering necroptosis and pyroptosis
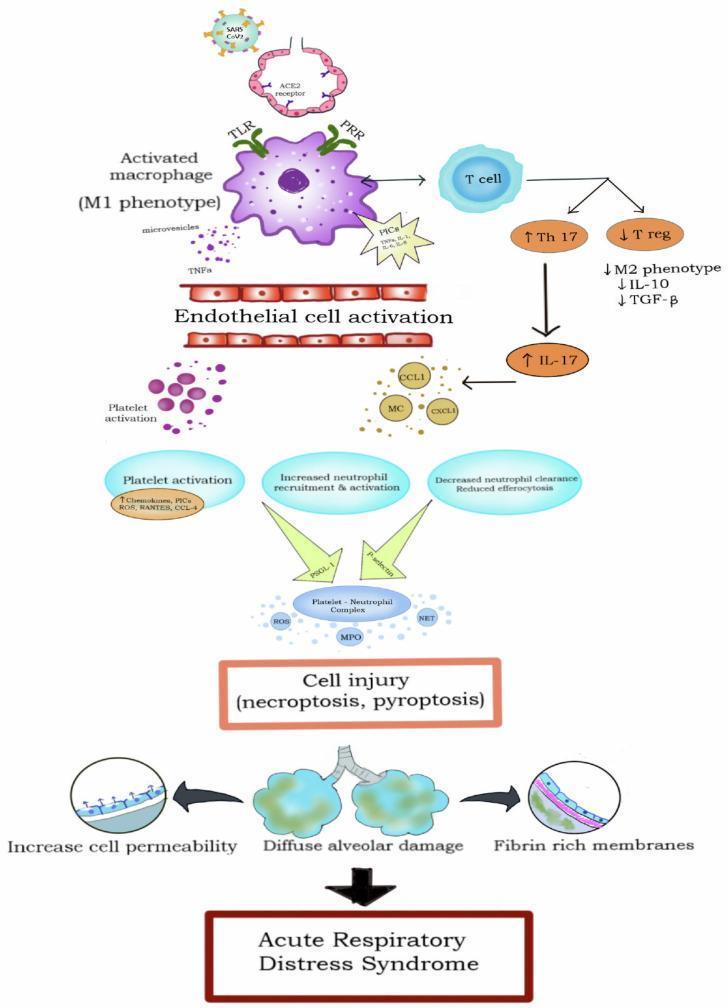
3c. Cytokine Storm / Inflammatory Amplification
A self-amplifying inflammatory cascade drives ongoing injury:
- Alveolar macrophages activated via TLR/PRR pathways polarize to the M1 (pro-inflammatory) phenotype
- They release pro-inflammatory cytokines: TNF-α, IL-1β, IL-6, IL-8 (CXCL8), IL-17
- These cytokines recruit and activate more neutrophils and promote endothelial cell activation
- IL-8 in particular is a potent neutrophil chemoattractant responsible for sequestration within the lung
- The Th17/T-reg imbalance (↑Th17, ↓T-reg) further skews the milieu toward inflammation
- Anti-inflammatory mediators (IL-10, TGF-β) are relatively suppressed
In pancreatitis-associated ARDS, phospholipase A₂, elastase, and lipase released systemically directly increase vascular permeability, and phospholipase A₂ enzymatically degrades surfactant. — Murray & Nadel's Textbook of Respiratory Medicine
3d. Surfactant Dysfunction
- Plasma proteins leaking into the alveolar space inhibit surfactant function
- The proportion of large (active) to small (inactive) surfactant aggregates is diminished due to decreased production and increased conversion
- Neutrophil elastase degrades surfactant protein A
- Net result: loss of surface tension reduction → microatelectasis and alveolar collapse
- Unlike neonatal RDS (where surfactant deficiency is the primary event), in adult ARDS the surfactant abnormality is secondary to barrier disruption — explaining why surfactant replacement trials in adults have failed
3e. Coagulation–Inflammation Interplay
- Activation of the coagulation cascade within the injured lung leads to intravascular fibrin deposition and microthrombi in pulmonary capillaries
- Fibrin also accumulates in alveolar spaces, contributing to hyaline membrane formation
- This causes vascular obliteration and contributes to pulmonary hypertension
- Simultaneous suppression of fibrinolysis (via elevated PAI-1) prevents clot resolution
3f. Impaired Alveolar Fluid Clearance (Na⁺/Water Transport)
- Active fluid reabsorption from the alveolar space is driven by Na⁺-K⁺-ATPase on type II pneumocytes and epithelial Na⁺ channels (ENaC)
- Injury to type II pneumocytes disrupts this active transport, preventing clearance of alveolar edema
- This is compounded by the fact that type II pneumocytes are also the source of surfactant synthesis
3g. Angiopoietin Dysregulation
- Angiopoietin-2 (Ang2) is elevated in sepsis and ARDS and acts as a competitive antagonist to Ang1, disrupting endothelial barrier integrity
- Ang2 promotes pulmonary vascular leak; exogenous Ang2 in animal models induces pulmonary leak reversed by Ang1
- Genetic variants in Ang2 are associated with higher plasma levels and increased ARDS risk
- The Ang2/Tie2 receptor axis is an emerging therapeutic target — Murray & Nadel's Textbook of Respiratory Medicine
4. Physiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + surfactant loss | Microatelectasis → ↓ functional residual capacity, ↓ lung compliance |
| Alveolar flooding (fluid-filled units) | Right-to-left shunting → profound hypoxemia (V/Q mismatch) |
| Microthrombi + vascular obliteration | ↑ Dead space ventilation → ↑ minute ventilation requirement |
| Hypoxic vasoconstriction + fibrin deposition | Pulmonary hypertension |
| Heterogeneous injury | "Baby lung" — only a small fraction of lung is available for ventilation |
5. Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
Mechanical ventilation, though life-saving, can perpetuate or worsen injury:
- Volutrauma: Large tidal volumes overdistend "normal" lung units (the heterogeneous ARDS lung funnels volume preferentially to compliant regions)
- Barotrauma: Excessive airway pressure → pneumothorax, pneumomediastinum
- Atelectrauma: Repetitive opening/closing of collapsed terminal units generates high shear stresses at the interface of aerated and collapsed tissue
- Biotrauma: Mechanical stretch activates local inflammatory cells, releasing cytokines and ROS that spill into the systemic circulation, contributing to multi-organ failure
This is the rationale for lung-protective ventilation (6 mL/kg tidal volume, plateau pressure ≤30 cmH₂O) as the standard of care. — Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine
Summary Flow
Inciting insult (sepsis, pneumonia, aspiration, trauma…)
↓
Activation of alveolar macrophages → Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration in lung → ROS, proteases, NETs, platelet-neutrophil complexes
↓
Injury to type I pneumocytes + microvascular endothelium
↓
↑ Alveolar-capillary permeability
↓
Protein-rich edema floods alveoli + Coagulation activation → Fibrin/hyaline membranes
↓
Surfactant dysfunction → Microatelectasis
↓
Impaired Na⁺ transport (type II pneumocyte injury) → Failed fluid clearance
↓
ARDS: Hypoxemia + ↓ Compliance + Pulmonary Hypertension + ↑ Dead Space
↓
±
Ventilator-Induced Lung Injury (perpetuates the cycle)
↓
Resolution (proliferative → fibrotic) OR Death
Key sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (Etiology and Pathogenesis of ARDS)
- Harrison's Principles of Internal Medicine, 22nd ed., Chapter 312
- Goldman-Cecil Medicine, Chapter 90/91
- Morgan & Mikhail's Clinical Anesthesiology, 7th ed.
This is a shared conversation. Sign in to Orris to start your own chat.