Metabolic acidosis - Etiology,causes,types, pathophysiology and management - act like a professor in Anesthesia and explain this topic to junior resident as a long answer question for MD final exam
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
I now have all the material I need. Let me compose the comprehensive professor-level answer.
Metabolic Acidosis: A Comprehensive Answer for MD Final Examination
From an Anesthesiologist's Perspective
INTRODUCTION
Metabolic acidosis is one of the most common and clinically significant acid-base disturbances encountered in the perioperative setting, the ICU, and the emergency department. As an anesthesiologist, you must not only recognize it promptly but understand its mechanisms, classify it correctly, and manage it decisively - because unrecognized or inadequately treated metabolic acidosis profoundly influences anesthetic drug pharmacology and patient outcomes.
Definition: Metabolic acidosis is characterized by:
-
Arterial pH < 7.35 (acidemia)
-
Primary decrease in serum bicarbonate [HCO3-] < 21 mEq/L
-
Compensatory decrease in PaCO2 (respiratory compensation via hyperventilation)
-
Barash, Cullen & Stoelting's Clinical Anesthesia 9e, p. 1151
PART 1: ETIOLOGY AND PATHOPHYSIOLOGY
The Underlying Mechanism
The body's pH is maintained by the Henderson-Hasselbalch relationship:
pH = pK + log ([HCO3-] / [0.03 × PaCO2])
Metabolic acidosis results from one of four fundamental mechanisms:
- Increased bicarbonate loss - gastrointestinal (diarrhea) or renal (RTA, carbonic anhydrase inhibitors)
- Decreased renal excretion of acid - renal failure, RTA
- Imbalance between production and consumption of endogenous acids - lactic acidosis, ketoacidosis
- Administration of exogenous acid - ammonium chloride, lysine/arginine HCl, massive 0.9% NaCl infusion
To classify the etiology correctly, you must first calculate the Anion Gap (AG).
PART 2: THE ANION GAP - THE CORNERSTONE OF CLASSIFICATION
The plasma must maintain electrical neutrality. The anion gap represents the difference between "measured" cations and "measured" anions, which reflects unmeasured anions:
AG = [Na+] - ([Cl-] + [HCO3-]) Normal range = 7 to 14 mEq/L (commonly cited as ~12 mEq/L)
Using normal values: AG = 140 - (104 + 24) = 12 mEq/L
- Morgan & Mikhail's Clinical Anesthesiology 7e, p. 2227
Clinically important points about the AG:
- Plasma albumin accounts for the largest fraction (~11 mEq/L). For every 1 g/dL fall in albumin, the AG decreases by 2.5 mEq/L - so always correct the AG for hypoalbuminemia (common in ICU patients)
- Corrected AG = Measured AG + 2.5 × (4 - albumin in g/dL)
- Values > 20 mEq/L are diagnostically highly significant for high-AG acidosis
- Values > 30 mEq/L almost always indicate high-AG acidosis
PART 3: TYPES AND CAUSES OF METABOLIC ACIDOSIS
Metabolic acidosis is divided into two major categories based on the AG:
TYPE 1: HIGH ANION GAP (HAGMA) - "Unmeasured Anions Accumulate"
In HAGMA, the fall in HCO3- is replaced by accumulation of unmeasured anions (the conjugate bases of the offending acids). The classic mnemonic is:
MUDPILES
| Letter | Cause |
|---|---|
| M | Methanol |
| U | Uremia (advanced CKD) |
| D | Diabetic ketoacidosis (DKA), also Alcoholic & Starvation ketoacidosis |
| P | Paraldehyde / Propylene glycol |
| I | Isoniazid / Iron |
| L | Lactic acidosis |
| E | Ethylene glycol |
| S | Salicylates |
- Rosen's Emergency Medicine, Key Concepts p. 1413
A. Lactic Acidosis (Most Common in Anesthesia/ICU Setting)
Lactic acid is the end product of anaerobic glycolysis. Normal serum lactate is < 1 mmol/L; values > 2 mmol/L constitute hyperlactatemia, and > 4 mmol/L is frank lactic acidosis.
Classification (Cohen-Woods Classification):
-
Type A Lactic Acidosis - due to tissue hypoperfusion or acute hypoxia (oxygen delivery to tissues is impaired):
- Hemorrhagic/hypovolemic shock
- Septic shock
- Cardiogenic shock
- Severe hypoxemia
- Mesenteric ischemia
- Grand mal seizures
- Type A is far more common and most relevant in the perioperative period
-
Type B Lactic Acidosis - oxygen delivery is not impaired, but oxidative phosphorylation is impaired:
- B1: Underlying disease (liver failure, malignancy, HIV)
- B2: Drugs/toxins - metformin (very important!), propofol infusion syndrome, linezolid, antiretrovirals, cyanide poisoning
- B3: Inborn errors of metabolism (rare)
-
National Kidney Foundation Primer 8e, p. 1479; Brenner & Rector's The Kidney p. 652
B. Diabetic Ketoacidosis (DKA)
Insulin deficiency leads to unrestrained lipolysis. Free fatty acids are converted to ketone bodies (acetoacetate, beta-hydroxybutyrate) in the liver. These accumulate and consume HCO3-.
Remember: In DKA, phosphate is driven intracellularly. Serum phosphate may initially appear elevated but the total body deficit is significant. Treatment with insulin rapidly reverses hyperphosphatemia.
Specific treatment: insulin infusion, IV fluids (0.9% NaCl initially, then 0.45% NaCl), potassium replacement, phosphate, and magnesium.
C. Uremia (Renal Failure)
When GFR falls below 20 mL/min, the kidneys fail to excrete endogenously produced organic acids (sulfate, phosphate, urate, hippurate). These unmeasured anions accumulate, raising the AG.
D. Toxic Ingestions
- Methanol: Metabolized by alcohol dehydrogenase (ADH) to formaldehyde and then formic acid (highly toxic to retina). Symptoms delayed. Treatment: fomepizole or ethanol (competitive inhibitor of ADH), hemodialysis.
- Ethylene glycol: Metabolized to glycolic acid (principal cause of acidosis) and then oxalic acid, which deposits in renal tubules causing AKI.
- Salicylates: Mixed picture - direct respiratory stimulation causes respiratory alkalosis, while accumulation of acid intermediates causes metabolic acidosis. Treatment: urinary alkalinization with NaHCO3 to pH > 7.0 to trap ionized salicylate in urine and enhance elimination.
- Morgan & Mikhail's Clinical Anesthesiology 7e, p. 2228-2232
TYPE 2: NORMAL ANION GAP (NAGMA) / HYPERCHLOREMIC METABOLIC ACIDOSIS
In NAGMA, the fall in HCO3- is replaced by a rise in plasma [Cl-] (hence "hyperchloremic"). The unmeasured anion burden does not increase.
The mnemonic is HARDUP:
| Letter | Cause |
|---|---|
| H | Hyperalimentation / Hospital-acquired saline (0.9% NaCl) |
| A | Acid infusion / Addison's disease / Acetazolamide |
| R | Renal Tubular Acidosis (RTA) |
| D | Diarrhea |
| U | Ureterosigmoidostomy |
| P | Pancreatic fistula / drainage |
- Rosen's Emergency Medicine, p. 1417
A. Diarrhea
Diarrheal fluid contains 20-50 mEq/L of HCO3-. Loss of large volumes results in HCO3- depletion with compensatory Cl- retention. This is the most common cause worldwide.
B. Renal Tubular Acidosis (RTA)
RTA is a syndrome of systemic acidosis with inappropriately high (alkaline) urine pH relative to the systemic acidemia - the kidneys cannot adequately acidify the urine.
| Type | Defect | Urine pH | Serum K+ | Notes |
|---|---|---|---|---|
| Type 1 (Distal RTA) | Defective H+ secretion in distal tubule | > 5.5 | Low (hypokalemia) | Nephrocalcinosis common |
| Type 2 (Proximal RTA) | Defective HCO3- reabsorption in proximal tubule | Variable (< 5.5 once threshold exceeded) | Low | Fanconi syndrome |
| Type 3 | Combined defect (rare) | > 5.5 | Low | Carbonic anhydrase II mutation |
| Type 4 | Hypoaldosteronism or aldosterone resistance | < 5.5 | High (hyperkalemia!) | Diabetic nephropathy, NSAIDs, ACE inhibitors |
- Morgan & Mikhail's Clinical Anesthesiology 7e, p. 2230
Diagnostic aid: Urine Anion Gap (UAG)
UAG = ([urine Na+] + [urine K+]) - [urine Cl-]
- Normally positive or near zero
- In diarrhea: negative (NH4+ excretion rises, increasing urine Cl-)
- In RTA: positive despite systemic acidosis (NH4+ secretion impaired)
C. Dilutional / Hyperchloremic Acidosis from 0.9% NaCl
Massive infusion of 0.9% saline dilutes HCO3- and provides a chloride load. This is an important perioperative consideration - prefer balanced crystalloids (lactated Ringer's, Plasma-Lyte) over normal saline for large-volume resuscitation.
"Hyperchloremic metabolic acidosis is a predictable consequence of intraoperative infusion of 0.9% saline."
- Barash Clinical Anesthesia 9e, citing Scheingraber et al., Anesthesiology 1999
PART 4: RESPIRATORY COMPENSATION
In a spontaneously breathing patient, metabolic acidosis triggers peripheral and central chemoreceptors, driving compensatory hyperventilation to reduce PaCO2 and restore pH.
Winter's Formula (Expected Respiratory Compensation):
PaCO2 (expected) = 1.5 × [HCO3-] + 8 ± 2 mmHg
Or equivalently:
PaCO2 = 0.7 × [HCO3-] + 20 ± 5 mmHg
- Barash Clinical Anesthesia 9e, p. 1150
If the measured PaCO2 is:
- Lower than expected → additional respiratory alkalosis (e.g., salicylate poisoning, anxiety, pain)
- Higher than expected → additional respiratory acidosis (e.g., COPD, sedation, neuromuscular disease)
As an anesthesiologist, this is critically important: A mechanically ventilated patient CANNOT self-compensate. If you do not set the ventilator to maintain an appropriate PaCO2, you lose the respiratory buffer and pH deteriorates further.
Kussmaul's breathing is the classic clinical sign of compensatory hyperventilation in severe metabolic acidosis - deep, labored, sighing respirations.
PART 5: PHYSIOLOGICAL CONSEQUENCES OF METABOLIC ACIDOSIS
Understanding the systemic effects is essential for anesthetic management:
Cardiovascular Effects
- Decreased myocardial contractility (direct depressant effect of H+ on cardiac myocytes)
- Vasodilation (peripheral vasodilation; impaired pressor response)
- Increased pulmonary vascular resistance
- Increased risk of arrhythmias - halothane is particularly arrhythmogenic in acidosis
- Decreased threshold for ventricular fibrillation
- Impaired response to catecholamines (endogenous and exogenous)
Respiratory Effects
- Compensatory hyperventilation (Kussmaul breathing)
- Dyspnea, increased work of breathing
- Right shift of oxyhemoglobin dissociation curve (Bohr effect) → increased O2 delivery to tissues (initially beneficial)
Metabolic and Electrolyte Effects
- Hyperkalemia: H+ ions move into cells in exchange for K+ moving out (approximately 0.6 mEq/L rise in K+ for each 0.1 unit fall in pH)
- Insulin resistance, impaired glucose utilization
- Bone demineralization in chronic acidosis (H+ buffered by bone carbonate)
CNS Effects
- Cerebral vasodilation (though paradoxical intracellular brain acidosis can occur)
- Obtundation, coma in severe cases
- Seizures (particularly in toxic ingestions)
Anesthetic Drug Implications
-
Opioids: Most are weak bases; acidosis increases the non-ionized fraction, facilitating CNS penetration and potentiating sedative and respiratory depressant effects
-
Volatile agents (halothane especially): Cardiovascular depression is exaggerated; arrhythmias more likely
-
Intravenous induction agents: Circulatory depressant effects are potentiated; any agent reducing sympathetic tone can unmask circulatory collapse
-
Succinylcholine: AVOID in acidotic patients with hyperkalemia - depolarization will further raise plasma [K+], risking fatal arrhythmia
-
Morgan & Mikhail's Clinical Anesthesiology 7e, p. 2233
PART 6: DIAGNOSTIC APPROACH
A systematic step-wise approach is essential:
Step 1: Obtain Simultaneous ABG and Electrolytes
The calculated HCO3- on ABG and measured HCO3- (total CO2) on electrolytes should agree within ±2 mmol/L. Discrepancy suggests lab error or sampling issues.
Step 2: Confirm Metabolic Acidosis
- pH < 7.35, HCO3- < 21 mEq/L, low PaCO2 (compensatory)
Step 3: Calculate the Anion Gap
- AG = Na - (Cl + HCO3-)
- Correct for albumin if hypoalbuminemic
Step 4: If High AG - Consider the Delta-Delta (ΔΔ ratio)
ΔΔ = (Measured AG - Normal AG) / (Normal HCO3- - Measured HCO3-)
- ΔΔ 0.4-0.8: Mixed HAGMA + NAGMA (e.g., diarrhea + DKA)
- ΔΔ 1-2: Pure HAGMA
- ΔΔ > 2: HAGMA + concurrent metabolic alkalosis (e.g., vomiting + DKA)
This is vital for not missing a coexisting disorder!
Step 5: If Normal AG - Calculate Urine Anion Gap
Helps distinguish RTA (positive UAG, NH4+ secretion impaired) from diarrhea/GI losses (negative UAG, NH4+ secretion intact).
Step 6: If High AG - Calculate Osmolar Gap
Osmolar gap = Measured serum osmolality - Calculated osmolality
Calculated Osm = 2[Na+] + Glucose/18 + BUN/2.8
Normal osmolar gap is < 10 mOsm/kg. An elevated osmolar gap + HAGMA = toxic alcohol ingestion (methanol or ethylene glycol) until proven otherwise.
PART 7: MANAGEMENT
Management follows two parallel tracks: (A) treat the underlying cause and (B) treat the acidemia itself when life-threatening.
A. Treat the Underlying Cause (Always Primary)
| Cause | Specific Treatment |
|---|---|
| Lactic acidosis | Restore perfusion, oxygen delivery; treat sepsis source |
| DKA | Insulin infusion, IV fluids, K+/PO4/Mg replacement |
| Uremia | Renal replacement therapy (hemodialysis/CRRT) |
| Methanol/Ethylene glycol | Fomepizole (or ethanol), hemodialysis |
| Salicylate | Urinary alkalinization (NaHCO3 to urine pH > 7.0), hemodialysis |
| Diarrhea | Treat underlying diarrhea; oral/IV alkali supplementation |
| RTA Type 1/2 | Oral sodium bicarbonate/potassium citrate |
| RTA Type 4 | Fludrocortisone (if hypoaldosteronism); sodium bicarbonate |
| Hyperchloremic (saline) | Switch to balanced salt solution |
B. Alkali Therapy (Sodium Bicarbonate)
This remains controversial but clinically widely practiced. Key principles:
When to consider NaHCO3:
- pH remains < 7.20 despite treatment of underlying cause
- Severe acidosis with hemodynamic compromise
- Consider at pH < 7.20 in general, and < 7.30 in AKI (BICAR-ICU trial guidance)
The BICAR-ICU Trial (Jaber et al.):
A landmark multicenter open-label RCT studying bicarbonate in severe metabolic acidemia (pH < 7.20) in critically ill patients. Key findings:
-
No overall difference in the composite endpoint of 28-day mortality + organ failure at day 7
-
Secondary analysis: Patients with AKI had better mortality outcomes with bicarbonate and were less likely to need renal replacement therapy
-
This justifies targeted use in AKI-associated severe acidosis
-
Barash Clinical Anesthesia 9e, p. 1152
Calculating NaHCO3 Dose:
NaHCO3 required = Base Deficit × Bicarbonate Space × Body Weight
Where Bicarbonate Space = 25%-60% body weight (varies with severity; use 30% as starting estimate)
Example: 70 kg patient with BD = -10 mEq/L, Bicarb Space = 30% NaHCO3 = 10 × 0.30 × 70 = 210 mEq In practice: give 50% of calculated dose (105 mEq), recheck ABG, then titrate
Or use fixed empiric dose: 1 mEq/kg bolus
- Morgan & Mikhail's Clinical Anesthesiology 7e, p. 2232
Complications of NaHCO3 therapy:
- Paradoxical intracellular acidosis: CO2 generated from HCO3- + H+ readily enters cells, worsening intracellular pH, especially when ventilation is inadequate
- Sodium overload / hypernatremia
- Volume overload
- Rebound metabolic alkalosis ("overshoot")
- Hypokalemia (K+ shifts back intracellularly as pH rises)
- Hypocalcemia (ionized Ca2+ decreases as pH rises)
Alternative Buffers:
- THAM (Tris-hydroxymethyl aminomethane / Trometamol): Does not generate CO2 during buffering; does not increase sodium load. Theoretically superior to NaHCO3 but no clinical trial evidence to support it.
- Carbicarb (sodium carbonate-bicarbonate mixture): Produces less CO2 than pure NaHCO3, but similarly unproven.
C. Ventilatory Management (Critical in Anesthesia)
In mechanically ventilated patients:
- Maintain the compensatory respiratory alkalosis - set respiratory rate and tidal volume to target PaCO2 in the low 30s mmHg
- Aim to restore pH toward 7.25-7.30 as a reasonable target
- If you allow PaCO2 to rise to "normal" (40 mmHg) in a patient with metabolic acidosis, the pH will fall precipitously
"If a metabolic acidosis is recognized while the patient is mechanically ventilated, then every effort should be made to maintain ventilatory compensation."
- Barash Clinical Anesthesia 9e, p. 1152
Permissive hypercapnia in ARDS: When lung-protective ventilation (low tidal volume) is used, a degree of respiratory acidosis is acceptable ("permissive hypercapnia"), but this must be carefully weighed when metabolic acidosis is already present - the combined acidosis can be profound.
D. Renal Replacement Therapy
For severe, refractory acidemia (especially uremic or toxic ingestion):
- Hemodialysis with bicarbonate dialysate
- CRRT in hemodynamically unstable patients
PART 8: ANESTHETIC CONSIDERATIONS - THE PERIOPERATIVE CONTEXT
As an anesthesiologist, you will commonly encounter metabolic acidosis in:
- Septic/hemorrhagic shock - lactic acidosis; require aggressive resuscitation before and during surgery
- Diabetic patients - DKA perioperatively; high-risk for starvation ketoacidosis even with brief fasting
- Major abdominal surgery - large-volume normal saline resuscitation causes iatrogenic hyperchloremic metabolic acidosis; use balanced crystalloids
- Cardiac surgery - post-bypass lactic acidosis from low cardiac output, hepatic hypoperfusion
- Trauma - the "lethal triad" of hypothermia, coagulopathy, and acidosis
Key perioperative management points:
- Correct acidosis before elective surgery if pH < 7.30
- Avoid succinylcholine if acidosis + hyperkalemia
- Reduce induction agent doses (potentiated CNS/cardiovascular depression)
- Maintain active ventilatory compensation during mechanical ventilation
- Choose balanced crystalloids over 0.9% NaCl for resuscitation
- Monitor lactate as a marker of resuscitation adequacy (target < 2 mmol/L)
- The "rule of 15" for quick bedside assessment: In metabolic acidosis, PaCO2 should approximately equal the last two digits of the pH (e.g., pH 7.25 → expected PaCO2 ~25 mmHg) - though Winter's formula is more rigorous
SUMMARY TABLE
| Parameter | High AG Metabolic Acidosis | Normal AG (Hyperchloremic) Metabolic Acidosis |
|---|---|---|
| Anion Gap | > 14 mEq/L | Normal (7-14 mEq/L) |
| Chloride | Normal | Elevated |
| Mechanism | Accumulation of unmeasured anions | HCO3- loss replaced by Cl- |
| Mnemonic | MUDPILES | HARDUP |
| Key examples | Lactic acidosis, DKA, uremia, toxic alcohols | Diarrhea, RTA, saline infusion, RTA |
| UAG | N/A | Negative (diarrhea) / Positive (RTA) |
| Osmolar gap | Elevated in toxic alcohols | Normal |
COMPENSATION FORMULA (Must Know for Exam)
Metabolic Acidosis:
Expected PaCO2 = 1.5 × [HCO3-] + 8 ± 2 (Winter's Formula) Or: PaCO2 = 0.7 × [HCO3-] + 20 ± 5 mmHg
This compensation begins within minutes and is maximal by 12-24 hours. It does not completely normalize pH (compensatory hyperventilation merely restores pH toward normal, not to 7.40).
REFERENCES (Authoritative Textbooks)
- Barash, Cullen & Stoelting's Clinical Anesthesia, 9th Edition, Chapter 16
- Morgan & Mikhail's Clinical Anesthesiology, 7th Edition, Chapter 50
- Harrison's Principles of Internal Medicine, 22nd Edition, Chapter 58
- Rosen's Emergency Medicine, 9th Edition, Chapter 113
- Brenner & Rector's The Kidney, Volume 1, Chapter 16
- National Kidney Foundation Primer on Kidney Diseases, 8th Edition
Professor's closing note to the junior resident:
When you encounter a patient with unexplained hemodynamic instability, altered consciousness, or tachypnea in the perioperative period, always check an ABG and electrolytes simultaneously. Calculate the anion gap first - it takes 10 seconds and tells you whether you are dealing with an accumulation problem (HAGMA) or a loss/dilution problem (NAGMA). Treat the cause, not just the number. Sodium bicarbonate is not a "fix" - it buys you time. And never, ever forget to set your ventilator appropriately to maintain compensation in a mechanically ventilated patient - failing to do so in a patient with metabolic acidosis is one of the most dangerous oversights in critical care anesthesia.
Make a comparison table of all RTA types with clinical features and management
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
I have all the material needed. Here is the comprehensive comparison table with full clinical and management details.
Renal Tubular Acidosis (RTA) - Complete Comparison
Quick Orientation
All four types of RTA produce normal anion gap (hyperchloremic) metabolic acidosis with an inappropriately high urine pH relative to the degree of systemic acidosis - except Type 4, where the key distinguishing feature is hyperkalemia and urine pH is often < 5.5.
MASTER COMPARISON TABLE
| Feature | Type 1 (Distal) | Type 2 (Proximal) | Type 3 (Mixed) | Type 4 (Hyperkalemic Distal) |
|---|---|---|---|---|
| Primary Defect | Failure of H+ secretion in the distal tubule / collecting duct (type A intercalated cells) | Failure to reabsorb filtered HCO3- in the proximal tubule (threshold defect) | Combined proximal HCO3- wasting + distal H+ secretion defect | Hypoaldosteronism or aldosterone resistance → impaired H+ and K+ secretion in cortical collecting duct (CCD) |
| Tubular Site | Distal tubule / Cortical & Medullary Collecting Duct | Proximal tubule (S1-S3 segments) | Both proximal + distal | Cortical collecting duct (CCD) |
| Molecular Mechanism | Defect in: H+-ATPase (V-type), H+/K+-ATPase, or basolateral AE1 (Cl-/HCO3- exchanger) - all impair H+ secretion into lumen | Defect in: Na+/H+ exchanger (NHE3), carbonic anhydrase II/IV, or Na+-HCO3- cotransporter (NBCe1) - impairs proximal HCO3- reabsorption | Defect in carbonic anhydrase II (CA2 gene) - affects both segments | Deficiency of aldosterone or resistance to its action → decreased Na+ reabsorption → reduced electronegativity driving H+ and K+ secretion |
| Anion Gap | Normal | Normal | Normal | Normal |
| Serum HCO3- | < 15 mEq/L (can be very low, progressive if untreated) | 15-18 mEq/L (reaches a stable nadir; not progressive in steady state) | Very low (< 15 mEq/L) | Mild reduction: 18-22 mEq/L (rarely severe) |
| Serum K+ (KEY!) | Low (Hypokalemia) | Low (Hypokalemia, worsens with NaHCO3 treatment) | Low | HIGH (Hyperkalemia) - the hallmark |
| Urine pH | > 5.5 (unable to acidify urine even in systemic acidosis - cannot lower below 5.5) | Variable - alkaline (> 5.5) when above HCO3- threshold; < 5.5 when below threshold (i.e., in steady-state acidosis WITHOUT treatment) | > 5.5 | Usually < 5.5 (in hypoaldosteronism - NH3 is the main problem, not H+ secretion per se); may be > 5.5 in structural CCD damage |
| Urine Anion Gap (UAG) | Positive (NH4+ excretion severely impaired - cannot trap NH3 at alkaline pH) | Positive (in steady state) but can be negative transiently when above threshold | Positive | Positive (NH4+ severely impaired - hyperkalemia inhibits NH3 synthesis) |
| FE HCO3- | < 5% (minimal bicarbonate wasting when NOT receiving NaHCO3) | > 10-15% (massive bicarbonaturia when serum HCO3- rises with treatment) | Elevated (combined) | < 5% |
| Serum Cl- | Elevated (hyperchloremia) | Elevated | Elevated | Elevated |
| Serum Phosphate | Normal (unless nephrocalcinosis causes secondary damage) | Low (phosphaturia from Fanconi syndrome) | Low | Normal |
| Nephrolithiasis / Nephrocalcinosis | YES - common and characteristic (hypercalciuria + hypocitraturia + alkaline urine → calcium phosphate stones; nephrocalcinosis is a reliable marker for distal RTA) | NO (Exception: topiramate use - inhibits renal CA, also causes hypercalciuria) | No (osteopetrosis is the feature instead) | No |
| Bone Disease | Osteomalacia / Rickets (acid-driven bone mineral dissolution; hypercalciuria) | Osteomalacia / Rickets (renal phosphate wasting; impaired 1,25-OH vitamin D synthesis) | Osteopetrosis (dense, brittle bones - the classical triad with CA-II mutation) | Rare; not a primary feature |
| Other Fanconi Features | Absent | Present (when generalized proximal dysfunction): glucosuria (without hyperglycemia), aminoaciduria, phosphaturia, uricosuria, proteinuria (low-MW) | Absent | Absent |
| Severity of Acidosis | Severe and progressive (no steady state) | Mild-moderate and non-progressive (reaches a new steady state at lower HCO3-) | Severe | Mild (HCO3- rarely < 18 mEq/L) |
| NH4+ Excretion | Very low (failure to trap NH3 in acidic lumen) | Low-normal (NH3 production intact; issue is bicarbonaturia) | Very low | Very low (hyperkalemia inhibits NH3 synthesis in proximal tubule) |
CAUSES TABLE
| Cause Category | Type 1 (Distal) | Type 2 (Proximal) | Type 4 (Hyperkalemic) |
|---|---|---|---|
| Primary / Genetic | Autosomal dominant/recessive (H+-ATPase mutations - B1 subunit with sensorineural deafness; a4 subunit without deafness; AE1 mutations) | Isolated proximal RTA: NBCe1 (SLC4A4) mutation with ocular abnormalities; NHE3 defect | Rare congenital hypoaldosteronism |
| Autoimmune / Systemic | Sjögren syndrome (most common acquired cause); SLE; primary biliary cirrhosis; hypergammaglobulinemia; rheumatoid arthritis | SLE, Sjögren | Adrenal destruction (Addison's disease) |
| Hereditary Metabolic | Wilson disease, hereditary fructose intolerance | Cystinosis (most common in children); Wilson disease; galactosemia; tyrosinemia; Lowe syndrome; hereditary fructose intolerance; glycogen storage disease type I | Congenital adrenal hyperplasia (21-hydroxylase deficiency) |
| Drugs / Toxins | Amphotericin B (backleak/gradient defect); lithium; toluene (glue sniffing); ifosfamide | Acetazolamide, topiramate (carbonic anhydrase inhibitors); aminoglycosides; cisplatin; ifosfamide; tenofovir; valproate; heavy metals (lead, mercury, cadmium); myeloma proteins | NSAIDs (suppress renin); ACE inhibitors / ARBs; heparin (high-dose); trimethoprim; pentamidine; amiloride, triamterene, spironolactone, finerenone (K-sparing diuretics); cyclosporine, tacrolimus |
| Structural / CKD | Interstitial nephritis; medullary sponge kidney; sickle cell nephropathy; renal transplant rejection | Multiple myeloma (most common acquired in adults) | Diabetes mellitus (most common overall - hyporeninemic hypoaldosteronism); CKD with interstitial disease; sickle cell nephropathy; obstructive uropathy; lupus interstitial nephritis |
| Endocrine | Hyperparathyroidism; hyperthyroidism | Vitamin D deficiency / deficiency of 1-alpha hydroxylase | Primary hypoaldosteronism; isolated hypoaldosteronism; pseudohypoaldosteronism type I/II |
| Other | Marfan syndrome (rare) | Heavy metal poisoning | Beta-blockers (decrease renin release) |
DIAGNOSTIC ALGORITHM TABLE
| Diagnostic Test | Type 1 | Type 2 | Type 3 | Type 4 |
|---|---|---|---|---|
| Serum K+ | Low | Low (mild; worsens with alkali Rx) | Low | HIGH |
| Urine pH at steady state | > 5.5 | < 5.5 (can acidify at steady state) | > 5.5 | Usually < 5.5 (mineralocorticoid defect); may be > 5.5 (structural) |
| Urine pH during oral NH4Cl load test | Cannot drop below 5.5 | Can drop below 5.5 | Cannot drop below 5.5 | Can sometimes drop below 5.5 |
| UAG = ([uNa+] + [uK+]) - [uCl-] | Positive | Positive (in steady state) | Positive | Positive |
| FE HCO3- with alkali loading | < 5% | > 10-15% | Elevated | < 5% |
| Plasma renin | Normal/High | Normal | Normal | Low (in hyporeninemic hypoaldosteronism - DM, NSAIDs) or High (in Addison's) |
| Plasma aldosterone | Normal | Normal | Normal | Low |
| Serum phosphate | Normal | Low (if Fanconi) | Low | Normal |
| Urine glucose | Absent | Present (euglycemic glycosuria if Fanconi) | Absent | Absent |
| Serum HCO3- nadir | < 15 mEq/L | 15-18 mEq/L | < 15 mEq/L | 18-22 mEq/L |
| Nephrocalcinosis on imaging | YES (key finding!) | No | No | No |
MANAGEMENT COMPARISON TABLE
| Management Aspect | Type 1 (Distal) | Type 2 (Proximal) | Type 3 (Mixed) | Type 4 (Hyperkalemic) |
|---|---|---|---|---|
| Primary Goal | Correct acidosis + prevent nephrocalcinosis/stones | Correct acidosis (difficult due to bicarbonaturia) | Correct acidosis | Treat hyperkalemia first - acidosis often corrects simultaneously |
| Alkali Dose Required | Low - 1 to 3 mEq/kg/day (small amounts sufficient because distal nephron can retain given HCO3-) | Very high - 10 to 15 mEq/kg/day (large amounts lost in urine as kidneys waste the supplemented HCO3-) | Moderate-high (combined) | Modest - 1 to 2 mEq/kg/day if needed |
| Alkali Form | Sodium bicarbonate tablets OR potassium citrate (preferred - also treats hypokalemia and reduces stone risk; citrate metabolized to HCO3-) | Sodium bicarbonate + thiazide diuretic (induces mild ECF volume contraction, raising proximal reabsorption and reducing bicarbonaturia) + potassium supplements (anticipate worsening hypokalemia) | Alkali supplementation; treat underlying CA-II deficiency | Sodium bicarbonate (small doses) AFTER controlling hyperkalemia |
| Treat Hypokalemia? | Yes - potassium citrate is the alkali of choice (serves dual purpose) | Yes - must supplement K+ with any alkali given (alkali worsens hypokalemia by shifting K+ into cells + increasing distal K+ loss) | Yes | NO - serum K+ is already HIGH; avoid K+-containing alkali |
| Avoid K+-containing salts? | No (beneficial) | No (necessary) | No | YES - give sodium bicarbonate only (avoid K-citrate, K-Lyte) |
| Treat Nephrocalcinosis / Nephrolithiasis? | Yes - potassium citrate reduces stone formation (urinary citrate chelates Ca2+); correction of acidosis reduces hypercalciuria; thiazides may also reduce calciuria | Not applicable (nephrocalcinosis doesn't occur) | Not applicable | Not applicable |
| Treat Hyperkalemia? | Not applicable | Not applicable | Not applicable | Primary treatment step: discontinue offending drugs (NSAIDs, ACE-I/ARB, K-sparing diuretics); dietary potassium restriction; furosemide or thiazide (increases K+ and H+ excretion); fludrocortisone 0.1-0.3 mg/day (if mineralocorticoid deficiency confirmed) |
| Mineralocorticoid replacement? | No | No | No | Yes - fludrocortisone in confirmed hypoaldosteronism (use with caution in DM/HTN due to fluid retention risk) |
| Vitamin D / Phosphate supplementation? | Vitamin D if osteomalacia (treat acidosis first) | Phosphate supplementation + calcitriol if Fanconi syndrome present (phosphate wasting + impaired 1,25-OH vitamin D synthesis) | Phosphate + vitamin D | Not typically needed |
| Treat underlying disease | Sjögren syndrome, autoimmune - treat with immunosuppression where appropriate; stop amphotericin if possible | Stop offending drug (acetazolamide, tenofovir, etc.); treat myeloma; treat Wilson disease with chelation | Carbonic anhydrase II deficiency - supportive; bone marrow transplant has been tried | Stop causative drugs (NSAIDs, ACE-I, ARBs, K-sparing diuretics, heparin, calcineurin inhibitors, trimethoprim); treat DM; replace adrenal insufficiency |
| Alkali preparations available | Shohl's solution (1 mEq Na+/mL), NaHCO3 tablets (325 mg = 3.9 mEq; 650 mg = 7.8 mEq), K-citrate (Urocit-K), Polycitra-K | High-dose NaHCO3; potassium bicarbonate; thiazide diuretic added | High-dose alkali | Low-dose NaHCO3 only |
| Response to treatment | Excellent - acidosis corrects completely with adequate alkali; stones/nephrocalcinosis stabilize or improve; growth resumes in children | Partial - very large doses needed; treatment does not always fully correct acidosis | Partial | Excellent once hyperkalemia and the causative drug/disease is addressed |
SPECIAL CLINICAL PEARLS
Type 1 (Distal RTA) - "The Stone Maker"
- Most dangerous if untreated - progressive, severe acidosis
- Nephrocalcinosis is the pathognomonic complication (calcium phosphate deposits in medullary interstitium)
- Key triad: hypokalemia + urine pH always > 5.5 + nephrocalcinosis
- Amphotericin B causes a "gradient defect" - creates backleak channels in collecting duct membrane, allowing secreted H+ to leak back (urine pH may even be > 5.5 here too, but it is a permeability defect)
- Autosomal recessive distal RTA with sensorineural deafness (B1 subunit H+-ATPase mutation) - remember this for genetics MCQs
- Glue sniffing (toluene): causes distal RTA by inhibiting collecting duct H+ secretion + organic acid production
Type 2 (Proximal RTA) - "The Bicarbonate Waster"
- Can reach a steady state (nadir HCO3- 15-18 mEq/L) - acidosis does not progress indefinitely because once serum HCO3- falls enough, all filtered HCO3- is reabsorbed by the proximal tubule
- Paradox of treatment: When you give NaHCO3, serum HCO3- rises above the reabsorption threshold → massive bicarbonaturia → worsening hypokalemia (K+ secretion rises distally) - so you must give potassium alongside alkali
- Fanconi syndrome: think of proximal Type 2 RTA whenever you see the combination of glycosuria without hyperglycemia + hypokalemia + hypophosphatemia + aminoaciduria
- Most common inherited cause in children = cystinosis; most common acquired cause in adults = multiple myeloma
Type 3 (Mixed/CA-II deficiency) - "The Rare One with Dense Bones"
- Caused by deficiency of carbonic anhydrase II (CA-II), expressed in both proximal and distal tubules
- Classical triad: osteopetrosis + cerebral calcification + renal tubular acidosis
- Dense, brittle bones that fracture easily despite being radiographically dense
- Autosomal recessive (carbonic anhydrase II gene on chromosome 8)
- Rare and clinically important for exams; management is supportive
Type 4 (Hyperkalemic Distal RTA) - "The Common One Nobody Expects to be RTA"
- Most common RTA in clinical practice (usually encountered in elderly diabetic patients on ACE inhibitors or NSAIDs)
- The key diagnostic clue: normal anion gap acidosis + hyperkalemia in a diabetic with CKD - think Type 4 before anything else
- Mechanism of acidosis: hyperkalemia inhibits NH3 synthesis in proximal tubule → less NH4+ available as urinary buffer → net acid excretion decreases → acidosis develops
- Urine pH is variable: < 5.5 in mineralocorticoid deficiency (H+ secretion partially intact, but ammonia deficient); > 5.5 in structural CCD damage
- Treating the hyperkalemia often cures the acidosis - no need for large alkali doses
- Fludrocortisone works well but must be used cautiously in diabetic patients with hypertension (causes Na+ and fluid retention)
VISUAL SUMMARY: Type 4 RTA Pathophysiology
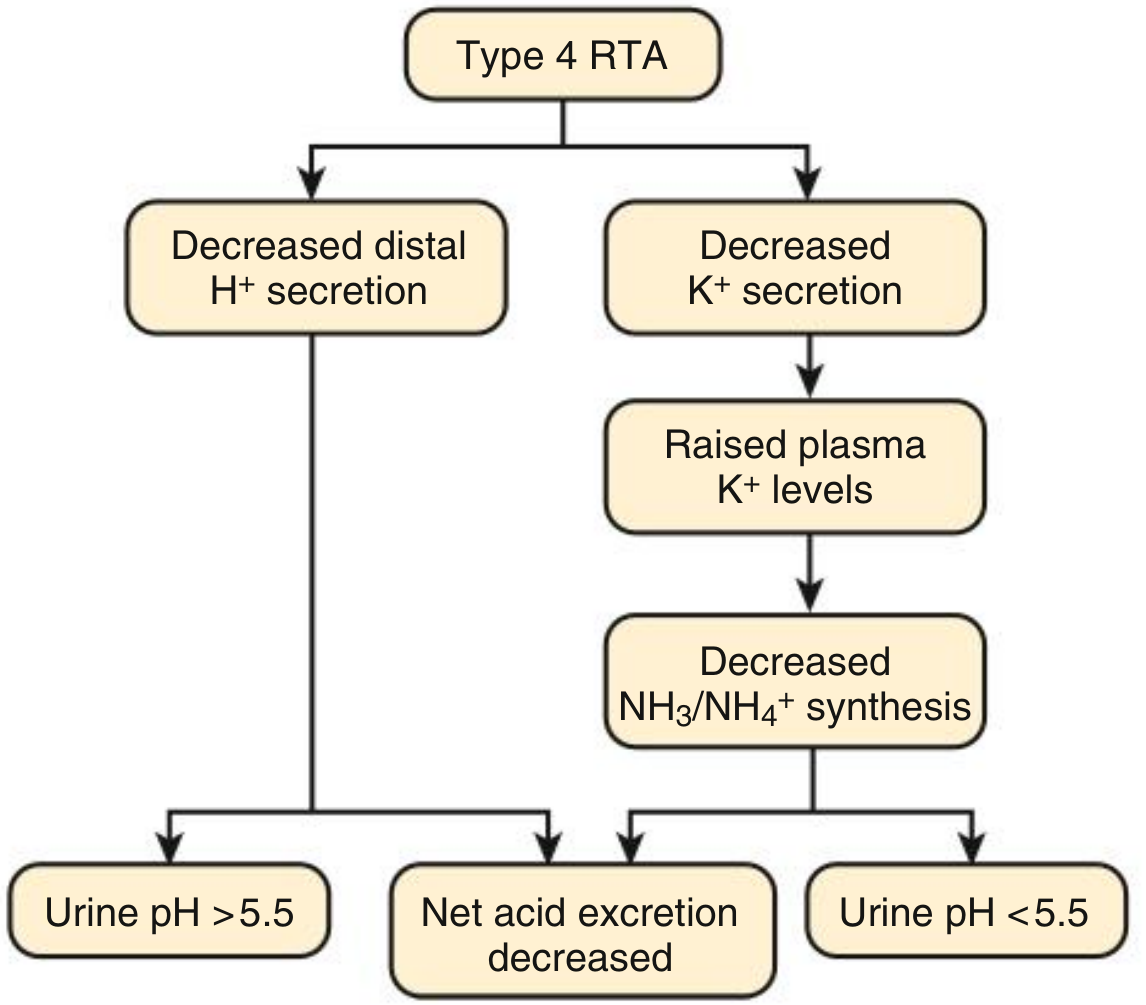
Fig: Urine pH in Type 4 RTA - net acid excretion is always reduced, but urine pH depends on whether the primary defect is in H+ secretion (urine pH > 5.5) or mineralocorticoid activity (urine pH < 5.5).
- Comprehensive Clinical Nephrology, 7th Edition, Fig. 13.3
ONE-LINE MEMORY HOOKS
| Type | One-line Hook |
|---|---|
| Type 1 - Distal | "Can't make acid urine, makes kidney stones instead - treat with low-dose potassium citrate" |
| Type 2 - Proximal | "Wastes bicarbonate in urine like a leaky bucket - needs enormous amounts of alkali plus K+ supplement" |
| Type 3 - Mixed | "CA-II mutation: stony bones + brain calcium + both tubule defects" |
| Type 4 - Hyperkalemic | "Diabetic on ACE inhibitor + high K+ + mild acidosis = fix the potassium, the acid fixes itself" |
Sources: Comprehensive Clinical Nephrology 7e pp. 197-205; Brenner & Rector's The Kidney pp. 169-442; Morgan & Mikhail's Clinical Anesthesiology 7e p. 2230; Murray & Nadel's Respiratory Medicine p. 2548-2606; National Kidney Foundation Primer on Kidney Diseases 8e
This is a shared conversation. Sign in to Orris to start your own chat.