Haemostasis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Now I have comprehensive material from Robbins Pathology. Let me compile the full answer.
Haemostasis
Haemostasis is the sequence of events by which the body arrests bleeding after vascular injury and then eventually dissolves the clot once healing is complete. It involves four overlapping components: vasoconstriction, primary haemostasis (platelet plug), secondary haemostasis (coagulation cascade/fibrin clot), and fibrinolysis.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 120-124
- Bailey and Love's Short Practice of Surgery, 28th Edition, p. 2018
1. Vasoconstriction
Immediately after vessel injury, neurohumoral factors cause transient vasoconstriction, reducing blood flow to the site. This is the first and fastest response.
2. Primary Haemostasis - Platelet Plug Formation
Platelets are anucleate disc-shaped fragments shed from bone marrow megakaryocytes. They carry two types of granules:
- α-granules: fibrinogen, vWF, factors V & XI, PDGF, TGF-β, fibronectin, PF4
- Dense (δ) granules: ADP, ATP, Ca²+, serotonin, epinephrine
The sequence proceeds in five steps:
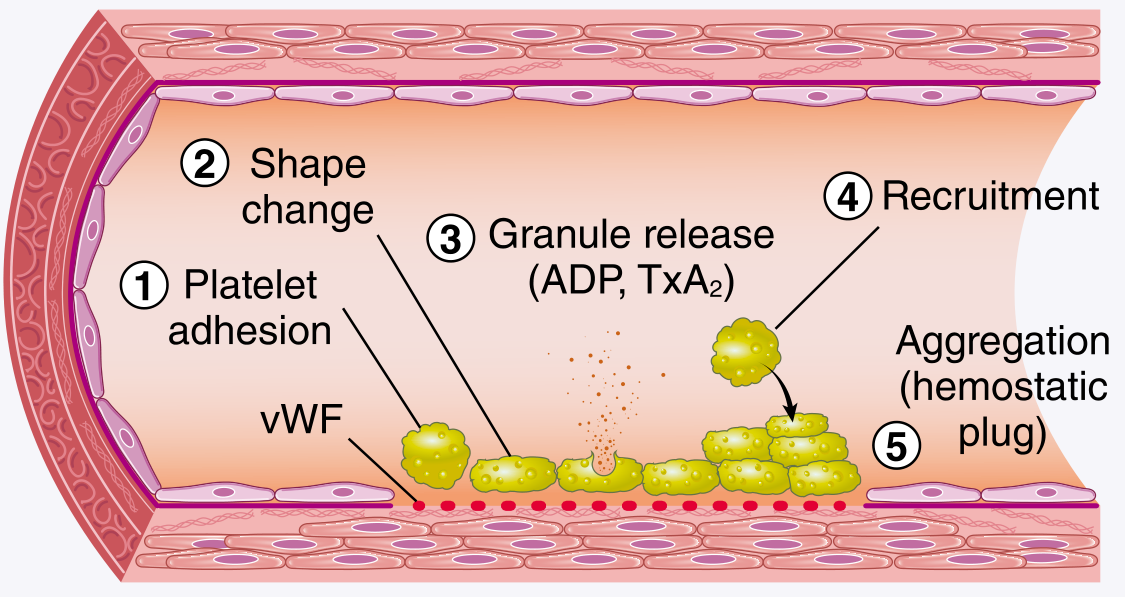
Step 1 - Platelet adhesion: After endothelial disruption, subendothelial collagen and von Willebrand factor (vWF) are exposed. Platelets bind via GpIb - vWF (especially under high shear) and via GpIa/IIa and GpVI - collagen. Deficiency of vWF = von Willebrand disease; deficiency of GpIb = Bernard-Soulier syndrome.
Step 2 - Shape change: Platelets convert from smooth discs to spiky "sea urchin" forms, greatly expanding surface area. Negatively charged phospholipids (phosphatidylserine) are translocated to the outer leaflet, acting as nucleation sites for coagulation factor complexes.
Step 3 - Granule release (secretion/activation): Triggered by thrombin (via PAR-1) and ADP. Dense granule ADP release begets further platelet activation (recruitment). Activated platelets also generate thromboxane A2 (TxA2) via cyclooxygenase - a potent aggregation inducer. Aspirin inhibits COX and thus TxA2 synthesis; P2Y12 antagonists (e.g. clopidogrel) block the ADP receptor.
Step 4 - Recruitment: ADP and TxA2 released from activated platelets recruit more platelets to the growing plug.
Step 5 - Aggregation: GpIIb/IIIa undergoes a conformational change, binding fibrinogen, which bridges adjacent platelets. Deficiency = Glanzmann thrombasthenia. The initial aggregation is reversible; thrombin-driven fibrin stabilisation makes it permanent.
3. Secondary Haemostasis - The Coagulation Cascade
The coagulation cascade amplifies the haemostatic response, generating an insoluble fibrin clot. Each step involves an enzyme (activated factor), a substrate (inactive proenzyme), and a cofactor, assembled on negatively charged phospholipid surfaces provided by activated platelets, in the presence of calcium.
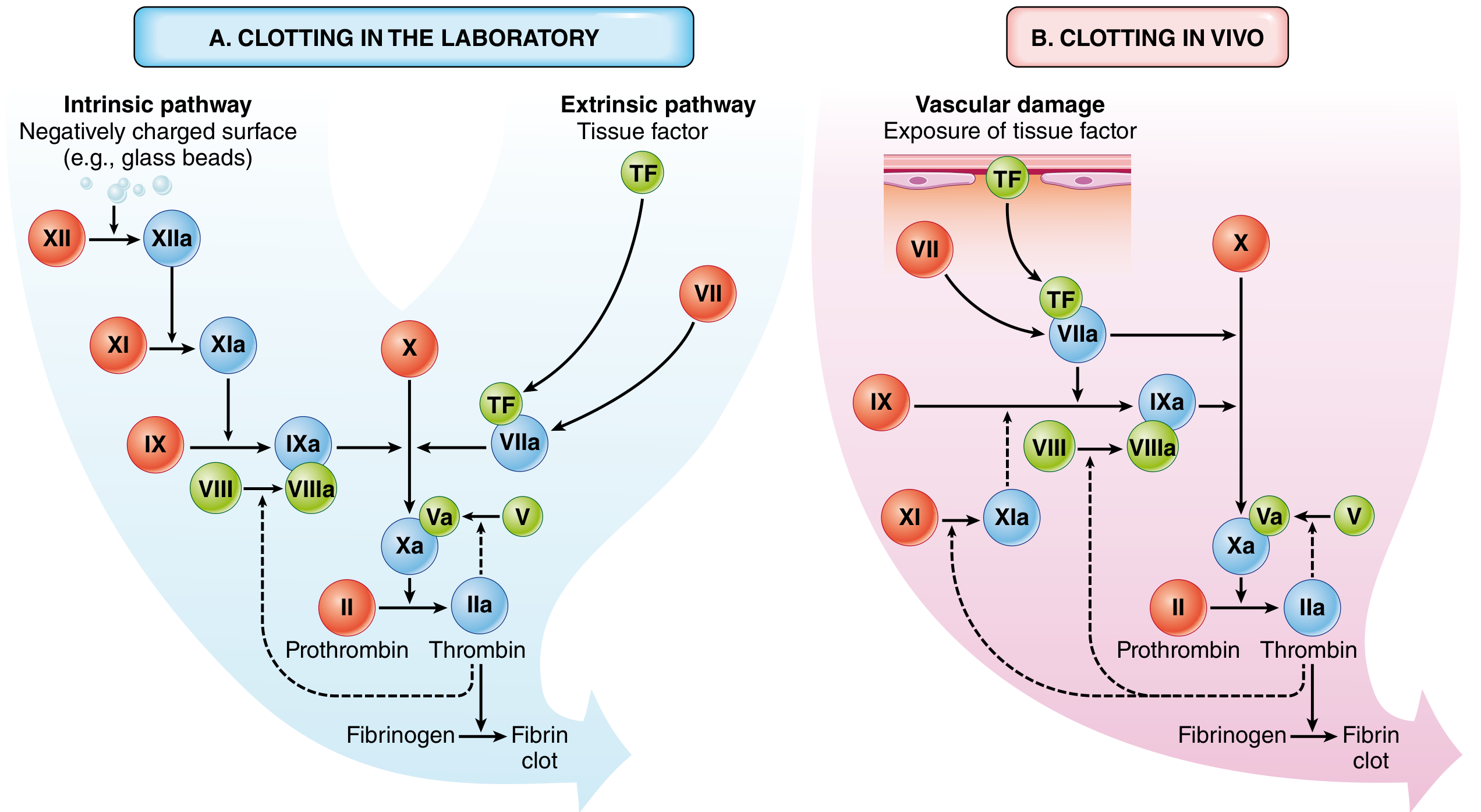
Extrinsic Pathway (in vivo initiator)
- Tissue factor (TF), constitutively expressed in the subendothelium, is exposed after injury
- TF + Factor VII → TF/VIIa complex
- TF/VIIa activates both Factor X (directly) and Factor IX (cross-talk with intrinsic pathway)
- Assessed clinically by the Prothrombin Time (PT)
Intrinsic Pathway (contact activation)
- Factor XII (Hageman factor) is activated by negatively charged surfaces
- XII → XIIa → XI → XIa → IX → IXa
- IXa + VIIIa (cofactor) → activates Factor X
- Assessed by Partial Thromboplastin Time (PTT)
Common Pathway
- Factor Xa + Va (prothrombinase complex) converts prothrombin (II) → thrombin (IIa)
- Thrombin cleaves fibrinogen → fibrin monomers, which polymerise
- Thrombin activates Factor XIII → XIIIa, which covalently cross-links fibrin strands, stabilising the clot
Note on clinical vs. in vivo: Factor XI deficiency causes only mild bleeding, meaning the intrinsic pathway plays a minor role in haemostasis in vivo. The TF/VIIa (extrinsic) complex is the dominant physiological initiator; factor IXa/VIIIa then amplifies it.
Vitamin K dependence: Factors II, VII, IX, and X require vitamin K for γ-carboxylation of glutamic acid residues, which allows calcium binding. Warfarin (coumadin) inhibits this step.
4. Thrombin - The Central Mediator
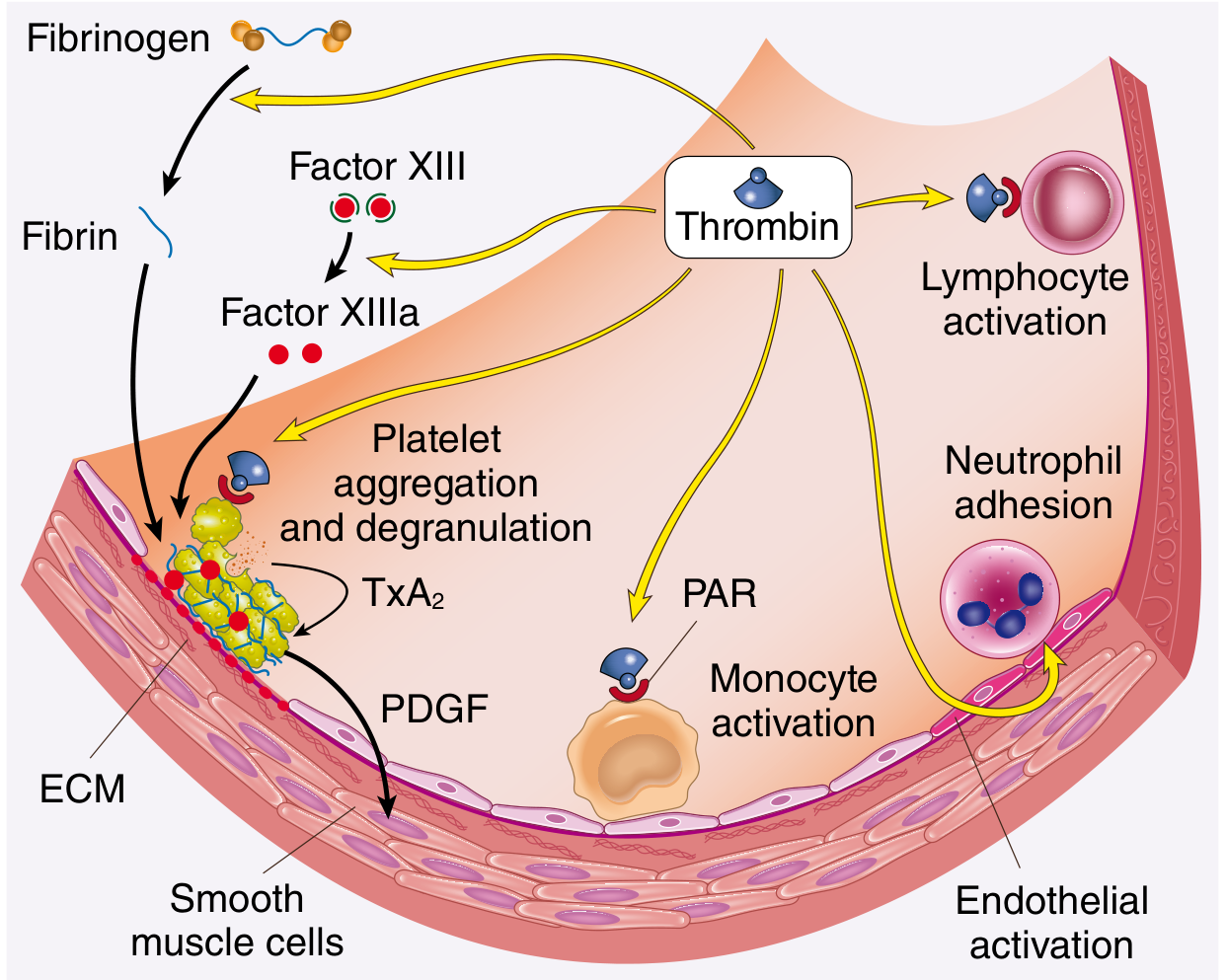
Thrombin is the most important coagulation factor, with roles beyond simple clot formation:
| Action | Effect |
|---|---|
| Cleaves fibrinogen | Generates cross-linked fibrin |
| Activates Factors V, VIII, XI | Amplifies the cascade (feedback) |
| Activates Factor XIII | Cross-links fibrin (clot stabilisation) |
| Activates PAR-1 on platelets | Induces platelet aggregation and degranulation |
| Activates PAR on endothelium, monocytes | Proinflammatory effects, tissue repair, angiogenesis |
| On normal endothelium | Switches to anticoagulant (via thrombomodulin) |
5. Limiting Coagulation - Natural Anticoagulants
Once initiated, coagulation must be confined to the injury site:
- Dilution - flowing blood washes away activated factors, cleared by the liver
- Phospholipid requirement - only activated platelets at the site provide the surface
- Endothelial anticoagulants (from intact endothelium adjacent to the clot):
- Prostacyclin (PGI2) and Nitric oxide (NO) - inhibit platelet activation and aggregation
- Adenosine diphosphatase - degrades ADP, removing the aggregation trigger
- Thrombomodulin - binds thrombin, switching it to activate protein C → inactivates factors Va and VIIIa
- Tissue factor pathway inhibitor (TFPI) - directly inhibits TF/VIIa and Xa
- Antithrombin III - inhibits thrombin, IXa, and Xa (potentiated by heparin)
6. Fibrinolysis - Clot Dissolution
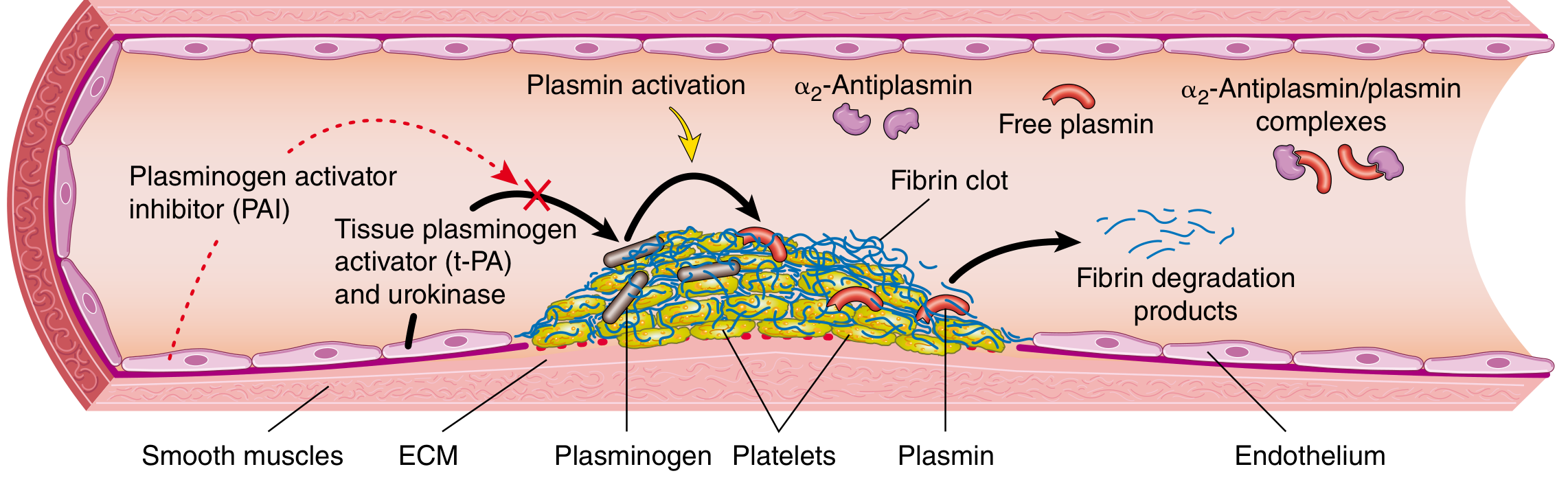
Once healing begins, the fibrin clot is dissolved by the fibrinolytic system:
- Plasminogen (inactive circulating precursor) accumulates within the clot
- Tissue plasminogen activator (t-PA) - released from endothelium, most active when bound to fibrin - converts plasminogen → plasmin
- Plasmin cleaves fibrin into fibrin degradation products (FDPs), including D-dimers (clinically useful marker of thrombosis, e.g. DVT/PE)
- α2-antiplasmin - inactivates free circulating plasmin, limiting systemic fibrinolysis
- Plasminogen activator inhibitor (PAI-1) - produced by endothelium in response to coagulation, dampens fibrinolysis
Summary Table: Key Haemostatic Factors
| Factor | Role | Deficiency |
|---|---|---|
| vWF | Platelet adhesion (GpIb bridge) | von Willebrand disease (↓ platelet adhesion + ↓ VIII carrier) |
| GpIb | Platelet adhesion receptor | Bernard-Soulier syndrome |
| GpIIb/IIIa | Fibrinogen receptor (aggregation) | Glanzmann thrombasthenia |
| Factor VIII | Intrinsic pathway cofactor | Haemophilia A |
| Factor IX | Intrinsic pathway enzyme | Haemophilia B (Christmas disease) |
| Factor VII | Extrinsic pathway (TF complex) | Prolonged PT only |
| Thrombin (IIa) | Fibrin, feedback, platelet activation | Incompatible with life if absent |
| Vitamin K | γ-carboxylation of II, VII, IX, X | Warfarin effect, ↑ PT and PTT |
| Protein C/S | Inactivate Va and VIIIa | Thrombophilia |
| Antithrombin III | Inhibits thrombin/IXa/Xa | Thrombophilia |
| t-PA | Fibrinolysis initiator | - |
| Plasmin | Fibrin degradation | - |
Clinical Tests
| Test | Pathway Assessed | Prolonged in |
|---|---|---|
| PT (INR) | Extrinsic (VII, X, V, II, fibrinogen) | Warfarin, liver disease, Vit K deficiency, Factor VII deficiency |
| APTT/PTT | Intrinsic (XII, XI, IX, VIII, X, V, II, fibrinogen) | Heparin, haemophilia A/B, lupus anticoagulant |
| Thrombin time | Fibrinogen → fibrin step | Fibrinogen deficiency, direct thrombin inhibitors |
| D-dimer | Fibrin degradation | DVT, PE, DIC (raised) |
| Platelet count | Primary haemostasis | Thrombocytopaenia |
| Bleeding time | Platelet function in vivo | Platelet disorders, vWD |
This is a shared conversation. Sign in to Orris to start your own chat.