Osteoclasts in rheumatology
Reading File
Finding Sources
Finding Sources
Searching PubMed
osteoclast AND rheumatoid arthritis AND bone erosion
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, well-illustrated content to write a thorough answer. Let me compile it.
Osteoclasts in Rheumatology
1. What Are Osteoclasts?
Osteoclasts are large, multinucleated cells derived from the fusion of mononuclear hematopoietic progenitor cells - specifically the granulocyte/macrophage progenitor (GMP, CFU-GM) lineage, the same precursor that gives rise to monocytes and granulocytes. They are not related to osteoblasts despite working alongside them. They attach to bone surfaces, create an acidic microenvironment via a ruffled border, and secrete proteolytic enzymes (cathepsin K, matrix metalloproteinases) to dissolve both the mineral and organic matrix of bone.
- Histology: A Text and Atlas, p. 600
2. The RANK-RANKL-OPG Axis: The Master Switch
The central molecular pathway governing osteoclast differentiation and activity is the RANK-RANKL-OPG system:
- RANK (Receptor Activator of NF-kB) is expressed on osteoclast precursors
- RANKL (RANK Ligand) is produced by osteoblasts, marrow stromal cells, and - critically in rheumatic disease - activated T lymphocytes
- OPG (Osteoprotegerin) is a soluble decoy receptor secreted by osteoblasts that binds RANKL and blocks it from engaging RANK
When RANKL binds RANK, it activates NF-kB signaling, promoting osteoclast differentiation and survival. When OPG binds RANKL first, it prevents this - acting as a natural brake on bone resorption. A second co-signal is M-CSF (macrophage colony-stimulating factor), also secreted by osteoblasts, which engages M-CSF receptors on precursors.
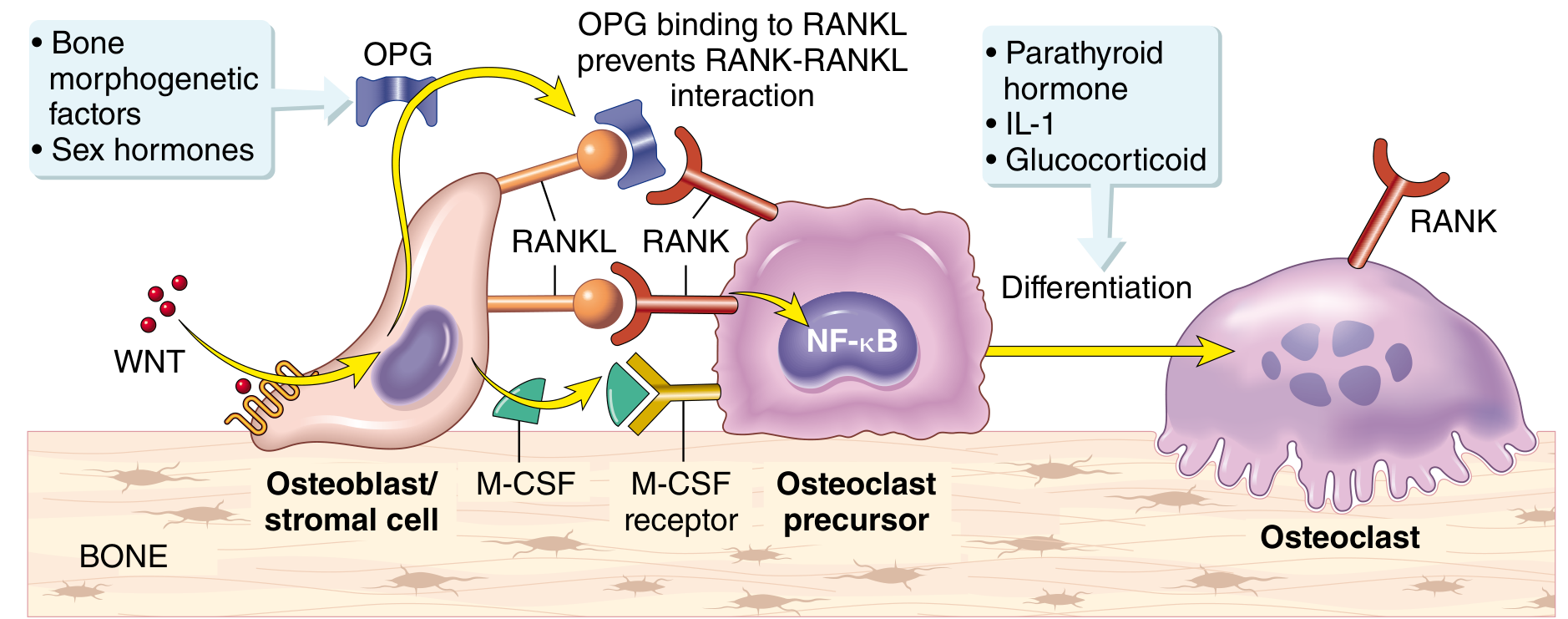
Paracrine mechanisms regulating osteoclast formation. RANKL binds RANK on osteoclast precursors activating NF-kB; OPG acts as a decoy receptor to inhibit this. WNT signaling promotes OPG production. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1082
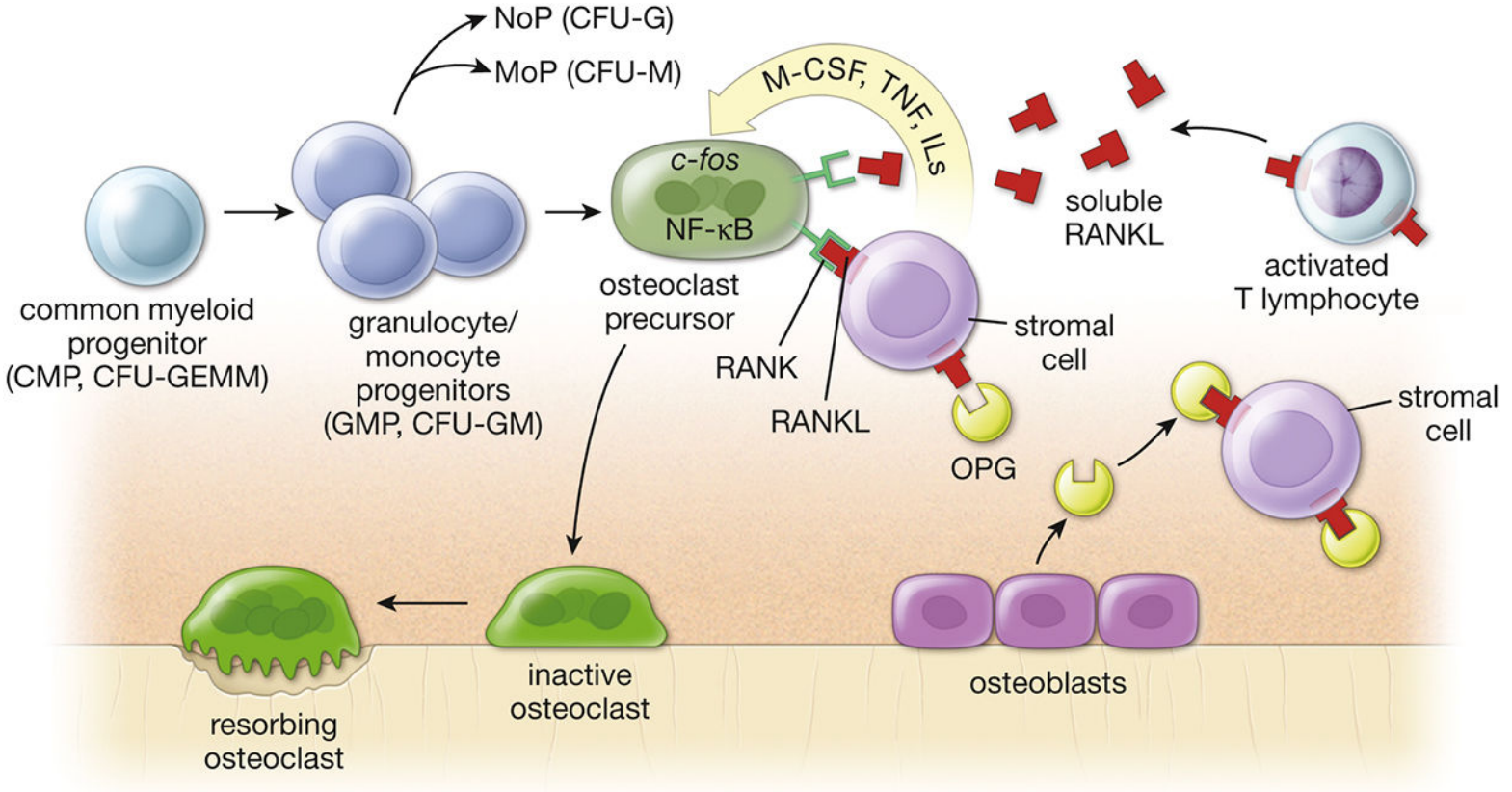
The origin of osteoclasts from GMP precursors. Note that activated T lymphocytes release soluble RANKL - a direct link to inflammatory/autoimmune disease. - Histology: A Text and Atlas, p. 600
3. Osteoclasts in Rheumatoid Arthritis (RA)
Bone erosion is a hallmark of RA, and osteoclasts are the primary mediators of this erosion. Goldman-Cecil Medicine states it directly:
"Bone damage requires cells with capacity to acidify the local milieu - osteoclast maturation and activation is a localized feature of rheumatoid arthritis synovium, arising as a consequence of RANKL, IL-1, TNF, and IL-17 activity. Thus activated osteoclasts localize in periarticular bone and in the adjacent bone marrow, thereby leading to the characteristic erosions detected on plain radiography."
- Goldman-Cecil Medicine, p. 2788
How the RA synovium drives osteoclast activation:
-
T cell activation: CD4+ T cells (Th1 and Th17) activated in the synovium produce both membrane-bound and soluble RANKL. This is a direct pathogenic link between adaptive immunity and bone destruction. Th17 cells also produce IL-17, a potent stimulator of RANKL expression.
-
Macrophage and synoviocyte cytokines: TNF-alpha, IL-1, and IL-6 from macrophages and fibroblast-like synoviocytes (FLS) further upregulate RANKL on stromal cells and suppress OPG, tipping the RANKL:OPG ratio in favor of osteoclastogenesis.
-
Autoantibody contribution: Rheumatoid factor and ACPAs (anti-citrullinated protein antibodies) - the seropositive markers of RA - may directly activate osteoclasts, in addition to activating macrophages via TLR/Fc receptor cross-talk. Seropositive RA patients have more severe erosive disease.
-
Pannus invasion: The inflammatory pannus tissue forms at the cartilage-pannus junction where FLS release MMPs destroying cartilage; osteoclasts simultaneously attack the underlying subchondral bone.
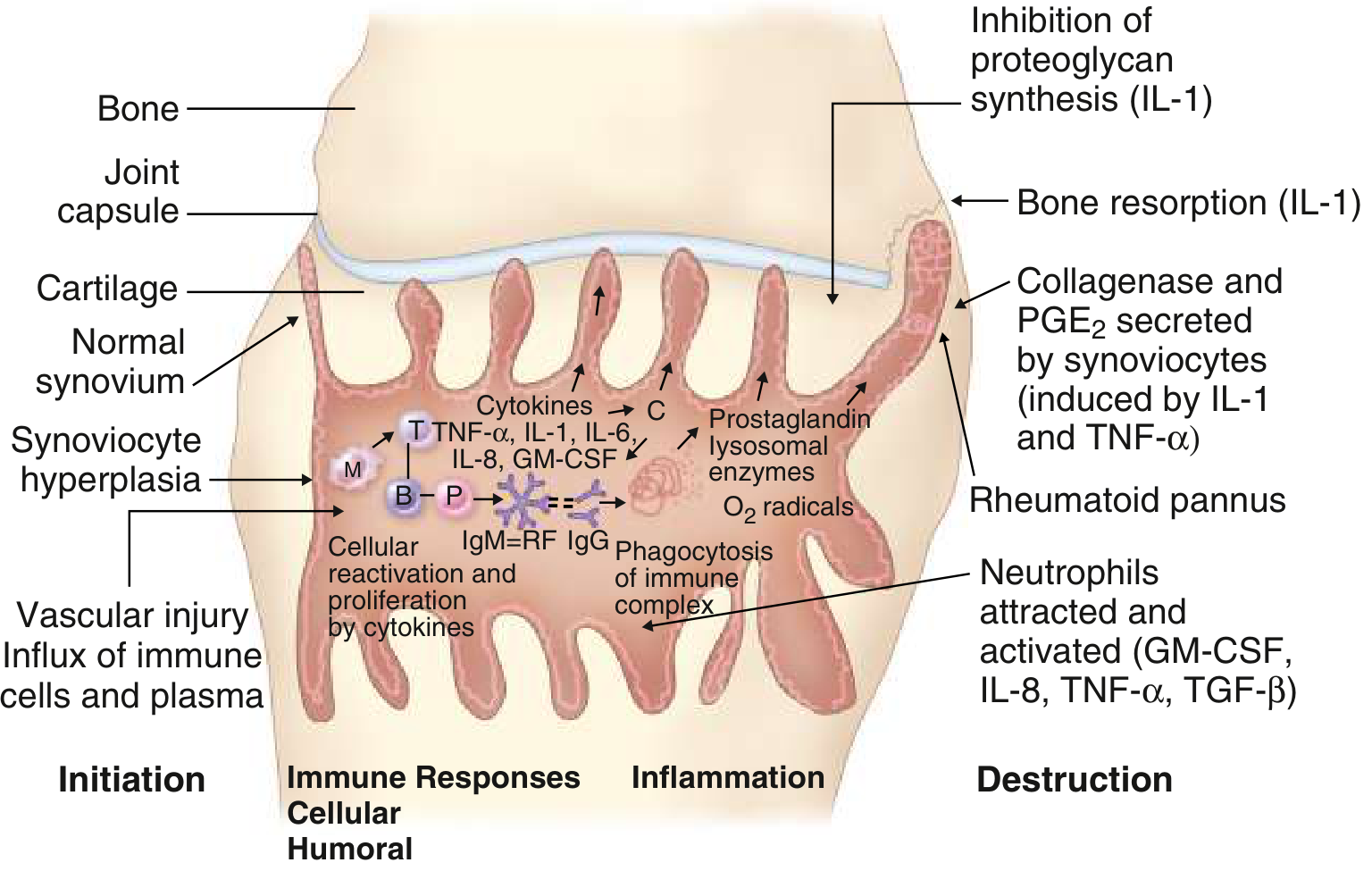
Events in RA synovitis from initiation through to bone resorption (IL-1) and destruction. B = B lymphocyte; M = macrophage; P = plasma cell; T = T lymphocyte; RF = rheumatoid factor. - Goldman-Cecil Medicine
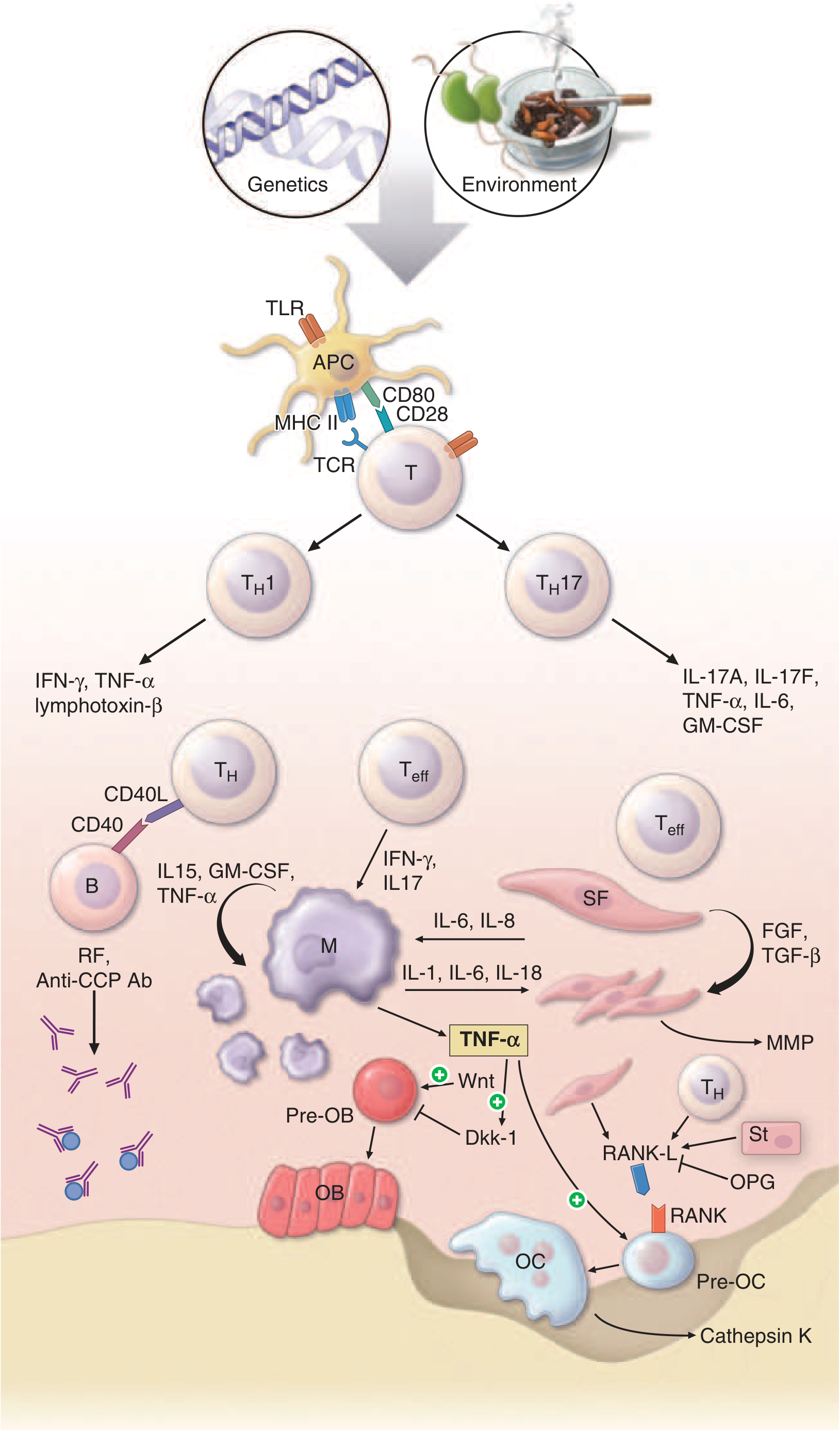
Pathophysiologic mechanisms of inflammation and joint destruction in RA. Note the RANKL/OPG axis at the bottom right, where T cells and stromal cells drive pre-osteoclast (Pre-OC) differentiation into active osteoclasts (OC) releasing cathepsin K. TNF-alpha also suppresses Wnt/Dkk-1 signaling, impairing osteoblast (OB) repair. - Harrison's Principles of Internal Medicine, 22nd ed.
4. Other Rheumatological Conditions
| Condition | Osteoclast Role |
|---|---|
| Psoriatic arthritis | Osteoclast-mediated erosion similar to RA, but paradoxically coexists with periosteal new bone formation |
| Ankylosing spondylitis | Less erosive; Wnt pathway is more active; syndesmophyte formation predominates over osteoclastic erosion |
| Systemic lupus (SLE) | Osteoclast-mediated resorption contributes to secondary osteoporosis; glucocorticoids used in treatment further upregulate RANKL |
| Gout/CPPD | Urate crystals activate macrophages to produce RANKL and IL-1, promoting periarticular osteoclast activity and tophi-associated bone erosions |
| Glucocorticoid-induced osteoporosis | Glucocorticoids directly upregulate RANKL and suppress OPG, accelerate osteoclastogenesis, and impair osteoblast function - a major iatrogenic concern in rheumatology |
5. Therapeutic Targeting of Osteoclasts
The RANK-RANKL-OPG axis is directly targeted by rheumatology-relevant therapies:
Denosumab
- A monoclonal antibody against RANKL
- Mimics the action of OPG - prevents RANKL from binding RANK, blocking osteoclast differentiation and activation
- Approved for glucocorticoid-induced osteoporosis and bone loss in RA
- A 2025 meta-analysis (PMID: 40335975) confirmed efficacy in RA-associated osteoporosis in RCTs
TNF Inhibitors (etanercept, adalimumab, infliximab)
- TNF-alpha is a major upstream driver of RANKL expression
- Blocking TNF reduces RANKL production and osteoclast activity
- Radiographic progression (erosion scores) is significantly reduced
IL-6 Inhibitors (tocilizumab)
- IL-6 upregulates RANKL on osteoblasts and T cells
- Tocilizumab reduces bone erosion scores and markers of bone resorption
Abatacept (CTLA-4-Ig)
- Blocks T cell co-stimulation (CD28-CD80/86 interaction)
- Reduces T cell activation and therefore T cell-derived RANKL production
JAK Inhibitors (tofacitinib, baricitinib, upadacitinib)
- Block signaling downstream of multiple cytokines (IL-6, IL-17, IFN-gamma) that drive RANKL expression
- Also inhibit osteoclastogenesis by blocking M-CSF receptor (CSF1R) signaling in some models
Bisphosphonates
- Inhibit osteoclast function by blocking farnesyl pyrophosphate synthase in the mevalonate pathway
- Widely used for glucocorticoid-induced osteoporosis in rheumatology patients
6. Regulatory Factors - Summary
| Stimulates Osteoclastogenesis | Inhibits Osteoclastogenesis |
|---|---|
| RANKL | OPG (osteoprotegerin) |
| M-CSF | Estrogen (suppresses RANKL) |
| TNF-alpha | Calcitonin |
| IL-1, IL-6, IL-17 | Bisphosphonates |
| PTH (prolonged exposure) | Denosumab |
| Glucocorticoids | Wnt/beta-catenin signaling |
| PGE2 | PTH (intermittent/pulsatile) |
- Histology: A Text and Atlas, p. 605; Robbins Pathologic Basis of Disease, p. 1082
Key takeaway: In rheumatological disease, osteoclasts are not incidental bystanders - they are the primary effectors of bone erosion. The inflammatory milieu of rheumatoid synovium hijacks the normal RANK-RANKL-OPG axis through T cell and cytokine-driven RANKL overexpression, converting the joint microenvironment into a site of accelerated, pathological bone resorption. Most modern DMARDs and biologics reduce bone erosion at least partly through indirect suppression of this pathway.
This is a shared conversation. Sign in to Orris to start your own chat.