drug distribution in bio pharmaceuticals
Reading File
Finding Sources
Finding Sources
Searching the Web
drug distribution biopharmaceutics volume of distribution protein binding tissue binding
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
I now have everything I need for a thorough answer. Let me compile the complete response.
Drug Distribution in Biopharmaceutics
Drug distribution is the process by which a drug reversibly leaves the bloodstream and enters extracellular fluid and tissues after absorption. It is the second phase of pharmacokinetics (ADME: Absorption, Distribution, Metabolism, Excretion).
1. Overview: What Happens After a Drug Enters the Blood?
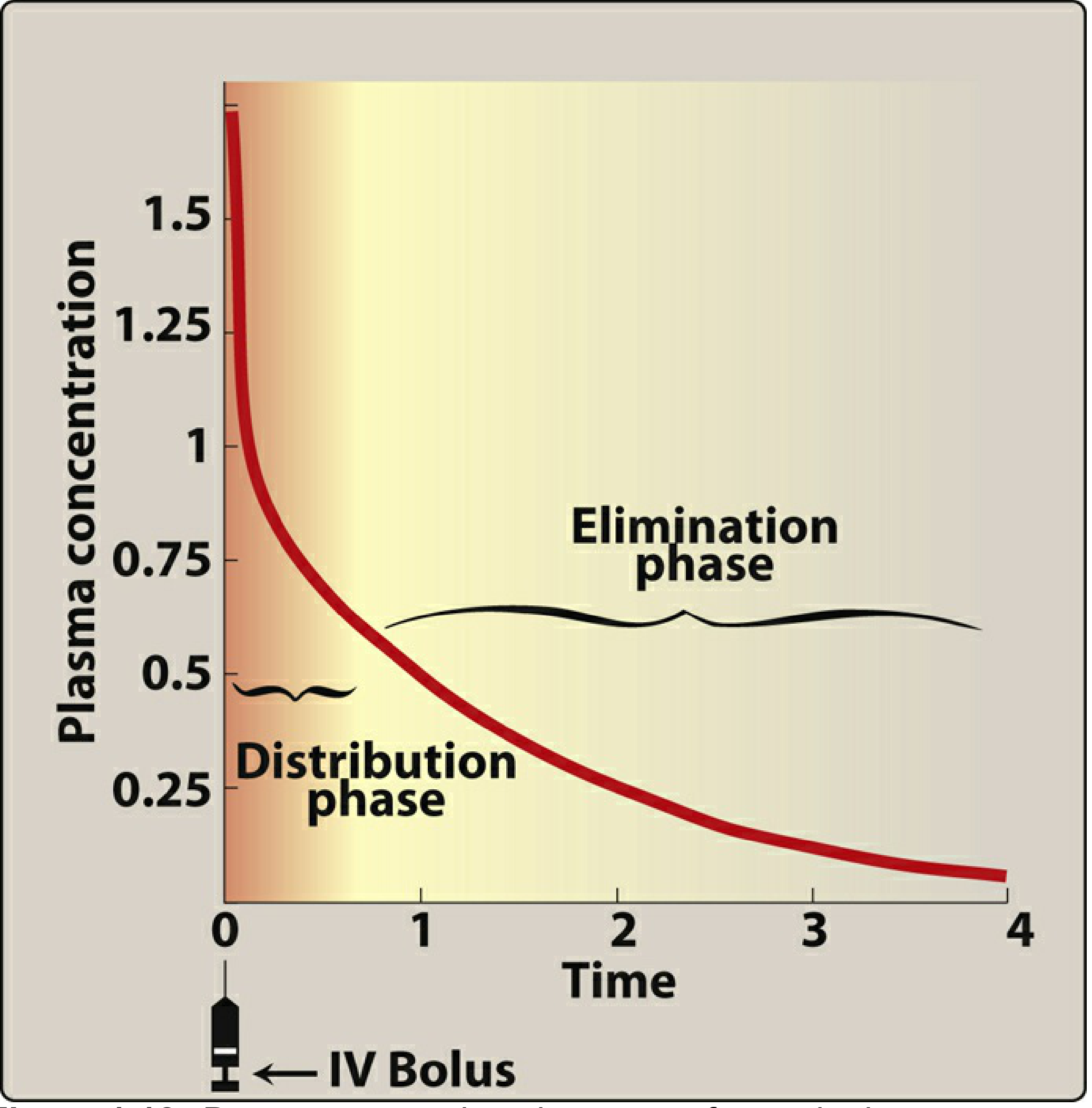
For an IV bolus dose, the plasma concentration-time curve shows two distinct phases:
- Distribution phase (alpha phase): Immediately after injection, plasma concentration falls rapidly as the drug moves out of blood into tissues.
- Elimination phase (beta phase): The slower, later decline as the drug is metabolized and excreted.
2. Factors Controlling Drug Distribution
A. Blood Flow (Perfusion)
The rate of drug delivery to a tissue depends on how well it is perfused. "Vessel-rich" organs - brain, liver, and kidney - receive high blood flow and equilibrate with drugs rapidly. Adipose tissue, skin, and resting muscle are low-perfusion tissues and equilibrate slowly.
Clinical example: Propofol is highly lipophilic and brain perfusion is high, so CNS drug levels rise rapidly, producing anesthesia within seconds. Once slower peripheral compartments (muscle, fat) absorb the drug, plasma concentration falls below the CNS equilibrium concentration, drug exits the brain, and consciousness returns. - Lippincott Illustrated Reviews: Pharmacology
B. Capillary Permeability
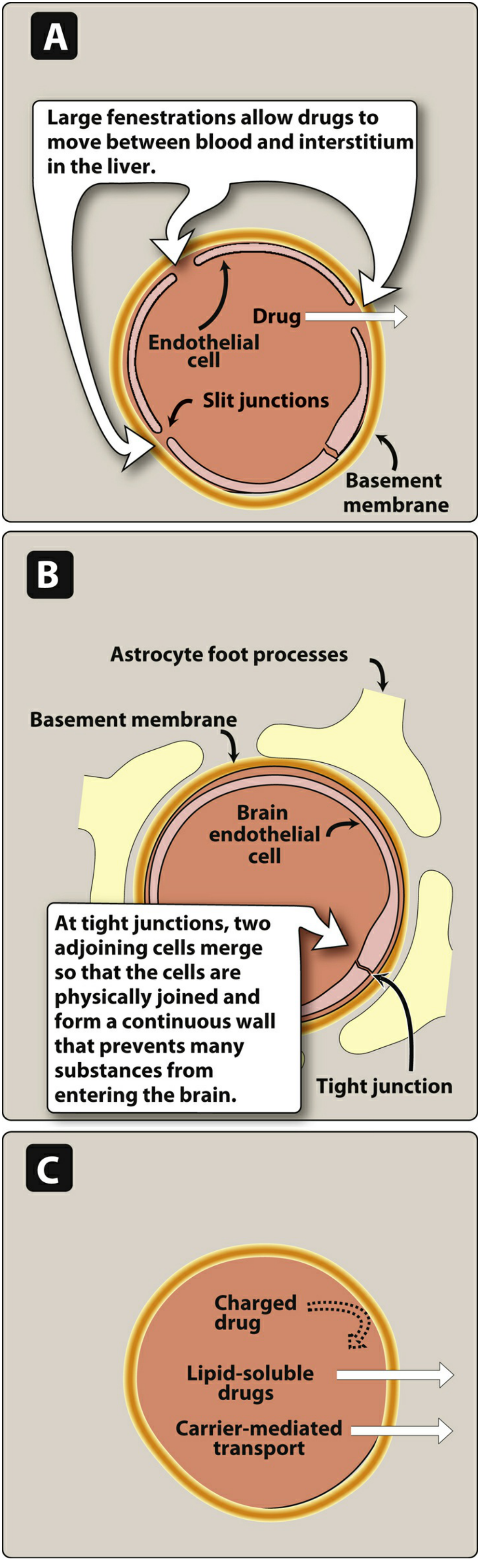
Capillary architecture varies across organs and governs drug penetration:
| Capillary Type | Location | Structure | Drug Access |
|---|---|---|---|
| Discontinuous (sinusoidal) | Liver, spleen, bone marrow | Large fenestrations, exposed basement membrane | Large proteins and drug-protein complexes pass freely |
| Fenestrated | Kidney, endocrine glands | Pores in endothelium | Small-medium molecules pass |
| Continuous (tight junctions) | Brain, testes, placenta | No slit junctions; astrocyte foot processes | Only lipid-soluble drugs, or carrier-mediated transport |
C. Plasma Protein Binding
Drugs in plasma exist in two forms:
- Bound (to plasma proteins) - pharmacologically inactive, too large to leave capillaries
- Free (unbound) - pharmacologically active, can diffuse into tissues
Key proteins:
- Albumin - binds acidic drugs (warfarin, phenytoin, salicylates, NSAIDs) and some neutral drugs
- Alpha-1-acid glycoprotein (AAG) - binds basic drugs (lidocaine, propranolol, some antidepressants)
- Lipoproteins - bind fat-soluble drugs
Key principle: Only the free drug crosses membranes, acts at receptors, and is metabolized/excreted. Protein-bound drug acts as a reservoir - as free drug is eliminated, bound drug dissociates to maintain the free fraction constant.
Drug displacement interactions: When two highly protein-bound drugs compete for the same binding site, one can displace the other, acutely raising free-drug levels and potentially causing toxicity - even with unchanged total plasma concentrations.
Special populations:
- In renal disease (CKD): Albumin levels fall and binding sites for acidic drugs (warfarin, phenytoin) are reduced - free drug fraction increases.
- In neonates: Protein binding is reduced for many drugs (diazepam, phenytoin, ampicillin). A drug that is 98% bound in adults may be only 90% bound in a neonate - free drug concentration rises 5-fold even at the same total concentration.
- Bilirubin competition: Drugs competing with bilirubin for albumin binding in jaundiced neonates can displace bilirubin into the brain, causing kernicterus (this is why sulfonamides are avoided in neonates). - Katzung's Basic and Clinical Pharmacology, 16e
D. Tissue Binding
Drugs accumulate in tissues when they bind to:
- Lipids - lipophilic drugs accumulate in adipose tissue (serves as long-duration reservoir; e.g., thiopental)
- Tissue proteins - some drugs bind muscle, bone, or organ proteins
- Nucleic acids - certain anticancer agents
Tissue reservoirs can prolong drug action (gradual release back to blood) or cause local toxicity (e.g., acrolein, the toxic cyclophosphamide metabolite, accumulates in the bladder and causes hemorrhagic cystitis). - Lippincott Illustrated Reviews: Pharmacology
E. Lipophilicity
Lipophilic (fat-soluble) drugs dissolve through lipid bilayer membranes and distribute widely. Hydrophilic (water-soluble) drugs are restricted to aqueous compartments and must cross via slit junctions or active transporters.
3. Volume of Distribution (Vd)
The apparent volume of distribution is the theoretical volume of fluid that would be required to contain the total amount of drug in the body at the same concentration as measured in plasma:
$$V_d = \frac{\text{Amount of drug in body}}{C_0 \text{ (plasma concentration at time 0)}}$$
It is not a real physiologic volume - it is a mathematical construct that tells you where the drug has "gone."
Three Main Distribution Compartments (in a 70-kg adult)
| Compartment | Approximate Volume | Vd of Drug | Drug Characteristics |
|---|---|---|---|
| Plasma | ~4 L | Low (~4 L) | High MW or highly protein-bound (e.g., heparin, warfarin) |
| Extracellular fluid (plasma + interstitium) | ~14 L | ~14 L | Low MW, hydrophilic (e.g., aminoglycosides) |
| Total body water | ~42 L | ~42 L | Low MW, lipophilic (e.g., ethanol) |
| Sequestered in tissues | Exceeds body volume | >100 L | High tissue affinity, low plasma levels (e.g., chloroquine ~15,000 L; digoxin ~667 L) |
Digoxin example: A 500 mcg dose in a 70-kg person yields a plasma concentration of ~0.75 ng/mL - giving a Vd of ~667 L, far exceeding total body volume. This is because digoxin is sequestered in muscle, adipose tissue, and bound to Na+/K+-ATPase, leaving very little in plasma. - Goodman & Gilman's Pharmacological Basis of Therapeutics
Chloroquine example: Vd ~15,000 L, reflecting extreme tissue sequestration in liver, spleen, and other organs.
Vd and Half-Life
$$t_{1/2} = \frac{0.693 \times V_d}{CL}$$
A larger Vd = longer half-life (more drug is "hidden" in tissues, unavailable for hepatic/renal clearance). A drug with high Vd takes longer to be fully eliminated because little circulates at any one time.
4. Special Barriers to Distribution
Blood-Brain Barrier (BBB)
- Brain capillaries have continuous tight junctions with no slit junctions and are surrounded by astrocyte foot processes.
- Only lipid-soluble, uncharged, low-MW drugs cross freely.
- Polar or ionized drugs require specific transporters (e.g., levodopa crosses via the large neutral amino acid transporter).
- P-glycoprotein (Pgp) efflux pumps actively expel many drugs back out of the CNS - this is a major barrier to CNS drug delivery. Neonates have less Pgp, making them more sensitive to morphine's CNS effects.
- In inflammation/meningitis, the BBB becomes more permeable, allowing drugs like penicillin (normally poorly penetrating) to reach therapeutic CSF concentrations.
Blood-Placenta Barrier
- Lipophilic drugs (alcohol, general anesthetics, many opioids) readily cross the placenta.
- The barrier is not absolute - teratogenicity is a major concern.
Blood-Testis Barrier
- Tight junctions between Sertoli cells protect germ cells from many drugs.
5. Special Populations Affecting Distribution
| Population | Effect on Distribution |
|---|---|
| Neonates/Infants | Higher total body water (75-85% of weight vs 50-60% in adults) - water-soluble drugs distribute into larger volume; less fat for lipid-soluble drugs; reduced protein binding |
| Elderly | Decreased lean body mass, increased fat percentage - lipophilic drugs have higher Vd; decreased albumin |
| Obesity | Increased fat mass - lipophilic drugs may have markedly elevated Vd |
| Renal disease (CKD) | Hypoalbuminemia, uremic toxins competing for binding sites - increased free fraction of acidic drugs |
| Liver disease | Reduced albumin synthesis, ascites (increased Vd of water-soluble drugs) |
6. Redistribution
Some drugs (notably barbiturates and propofol) undergo redistribution - they initially enter well-perfused tissues (brain, heart) rapidly, then slowly redistribute to less perfused fat and muscle. This redistribution terminates their clinical effect even before significant metabolism occurs, explaining the short duration of action of IV induction agents.
Summary Table
| Factor | Low Vd (stays in blood) | High Vd (distributes into tissues) |
|---|---|---|
| Protein binding | High (tightly bound to albumin) | Low (little plasma binding) |
| Lipophilicity | Low (hydrophilic) | High (lipophilic) |
| Molecular weight | High | Low |
| Tissue binding | Low | High |
| Examples | Heparin, warfarin | Chloroquine, amiodarone, digoxin |
Sources:
- Lippincott Illustrated Reviews: Pharmacology - Drug Distribution section
- Goodman & Gilman's The Pharmacological Basis of Therapeutics - Volume of Distribution
- Katzung's Basic and Clinical Pharmacology, 16e - Pediatric pharmacology and protein binding
- Goldman-Cecil Medicine - Volume of distribution and half-life
In 1o marks note for examination
Here is a concise, exam-ready 10-mark note on Drug Distribution in Biopharmaceutics:
Drug Distribution in Biopharmaceutics
(10-Mark Examination Note)
Definition
Drug distribution is the reversible transfer of a drug from the systemic circulation into tissues and body fluids after absorption. It is the "D" in the ADME pharmacokinetic model.
Factors Affecting Drug Distribution
1. Blood Flow (Perfusion)
- Well-perfused organs (brain, liver, kidney, heart) equilibrate rapidly.
- Poorly perfused tissues (fat, bone, resting muscle) equilibrate slowly.
- Example: Propofol reaches the brain rapidly due to high blood flow + high lipophilicity → rapid onset of anesthesia.
2. Capillary Permeability
| Capillary Type | Location | Drug Access |
|---|---|---|
| Discontinuous/Fenestrated | Liver, spleen | Large molecules pass freely |
| Continuous (tight junctions) | Brain, placenta, testes | Only lipid-soluble drugs or carrier-mediated drugs |
3. Plasma Protein Binding
- Drugs exist in plasma as bound (inactive, reservoir) and free/unbound (active) fractions.
- Albumin binds acidic drugs (warfarin, phenytoin, NSAIDs).
- Alpha-1-acid glycoprotein (AAG) binds basic drugs (lidocaine, propranolol).
- Only free drug crosses membranes, acts on receptors, and is eliminated.
- Drug displacement: Two drugs competing for albumin binding sites can displace each other → sudden rise in free drug → toxicity.
4. Tissue Binding
- Drugs bind to lipids, proteins, or nucleic acids in tissues.
- Creates a tissue reservoir that prolongs drug action.
- Example: Cyclophosphamide metabolite (acrolein) accumulates in bladder → hemorrhagic cystitis.
5. Lipophilicity
- Lipophilic drugs cross cell membranes freely; distributed widely.
- Hydrophilic drugs restricted to aqueous compartments; must use slit junctions or active transport.
Volume of Distribution (Vd)
$$\boxed{V_d = \frac{\text{Amount of drug in body}}{C_0 \text{ (plasma concentration at time zero)}}}$$
- A mathematical construct, not a real physiologic volume.
- Indicates where the drug has distributed relative to plasma.
Three Compartments (70-kg adult)
| Compartment | Volume | Vd of Drug | Example |
|---|---|---|---|
| Plasma only | ~4 L | ~4 L | Heparin, warfarin |
| Extracellular fluid | ~14 L | ~14 L | Aminoglycosides |
| Total body water | ~42 L | ~42 L | Ethanol, paracetamol |
| Tissue sequestration | >42 L (up to thousands) | Very high | Digoxin (~667 L), Chloroquine (~15,000 L) |
Vd and Half-Life Relationship
$$t_{1/2} = \frac{0.693 \times V_d}{CL}$$
- High Vd → long half-life (drug hidden in tissues, unavailable for elimination).
Blood-Brain Barrier (BBB)
- Brain capillaries have tight junctions - no slit junctions.
- Only lipid-soluble, low-MW, uncharged drugs cross freely.
- P-glycoprotein (Pgp) efflux pumps expel drugs back out of the CNS.
- Specific transporters carry polar drugs (e.g., levodopa via amino acid transporter).
- BBB becomes more permeable in meningitis (e.g., penicillin can reach therapeutic CSF levels).
Redistribution
- Drug moves from well-perfused organs (brain) to less-perfused tissues (fat, muscle) over time.
- Terminates drug effect before metabolism occurs.
- Example: Thiopental - short anesthesia due to redistribution, not rapid metabolism.
Special Populations
| Population | Effect |
|---|---|
| Neonates | More total body water (75-85%) → higher Vd for water-soluble drugs; less fat; reduced protein binding → more free drug → toxicity risk |
| Elderly | More fat, less lean mass → high Vd for lipophilic drugs; low albumin |
| Renal disease | Low albumin, uremic bilirubin competition → increased free fraction of acidic drugs |
| Obesity | Increased fat → very high Vd for lipophilic drugs |
Quick-Recall Points (for 10-mark answers)
- Distribution = reversible drug movement from blood → tissues
- Five factors: blood flow, capillary permeability, protein binding, tissue binding, lipophilicity
- Only free (unbound) drug is pharmacologically active
- Albumin binds acidic drugs; AAG binds basic drugs
- Vd = dose ÷ C₀; high Vd = extensive tissue distribution
- Digoxin Vd = 667 L (far exceeds body volume - extreme sequestration)
- BBB: tight junctions; only lipophilic/carrier-transported drugs enter
- t½ ∝ Vd - the more a drug distributes, the longer it persists
- Redistribution terminates action of IV anaesthetics (thiopental, propofol)
- Neonates at risk of toxicity due to reduced protein binding and immature BBB
Sources: Lippincott Illustrated Reviews: Pharmacology | Goodman & Gilman's Pharmacological Basis of Therapeutics | Katzung's Basic and Clinical Pharmacology, 16e
This is a shared conversation. Sign in to Orris to start your own chat.