Liver cirrhosis
Reading File
Finding Sources
Searching PubMed
"liver cirrhosis"[MeSH Terms] AND management
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Loading Image
Reading File
I now have comprehensive, rich content covering pathology, pathophysiology, complications, and scoring systems. Let me compile the full answer.
Liver Cirrhosis
Definition
Cirrhosis is the diffuse transformation of the liver into regenerative parenchymal nodules surrounded by fibrous bands. It is the morphologic end-stage of virtually any chronic liver disease — not a specific diagnosis, but a final common pathway with variable prognostic implications.
About 40% of individuals with cirrhosis are asymptomatic until the most advanced stages. — Robbins & Kumar Basic Pathology
Gross & Histological Appearance
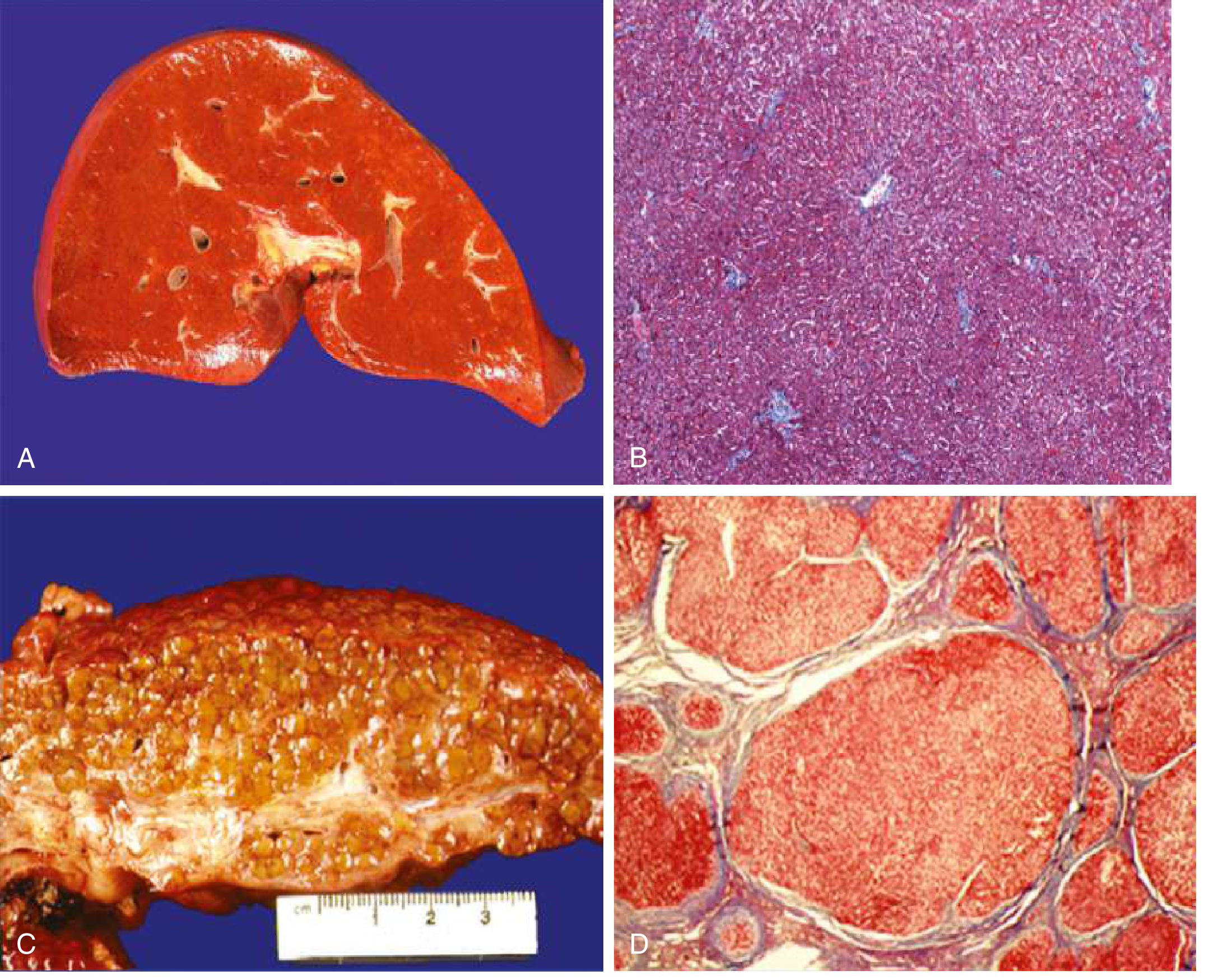
A/B: Normal liver with smooth surface and organized sinusoids. C/D: Cirrhotic liver — orange-tawny color, nodular surface, disorganized architecture with regenerative nodules surrounded by fibrous tissue (Masson's trichrome stain).
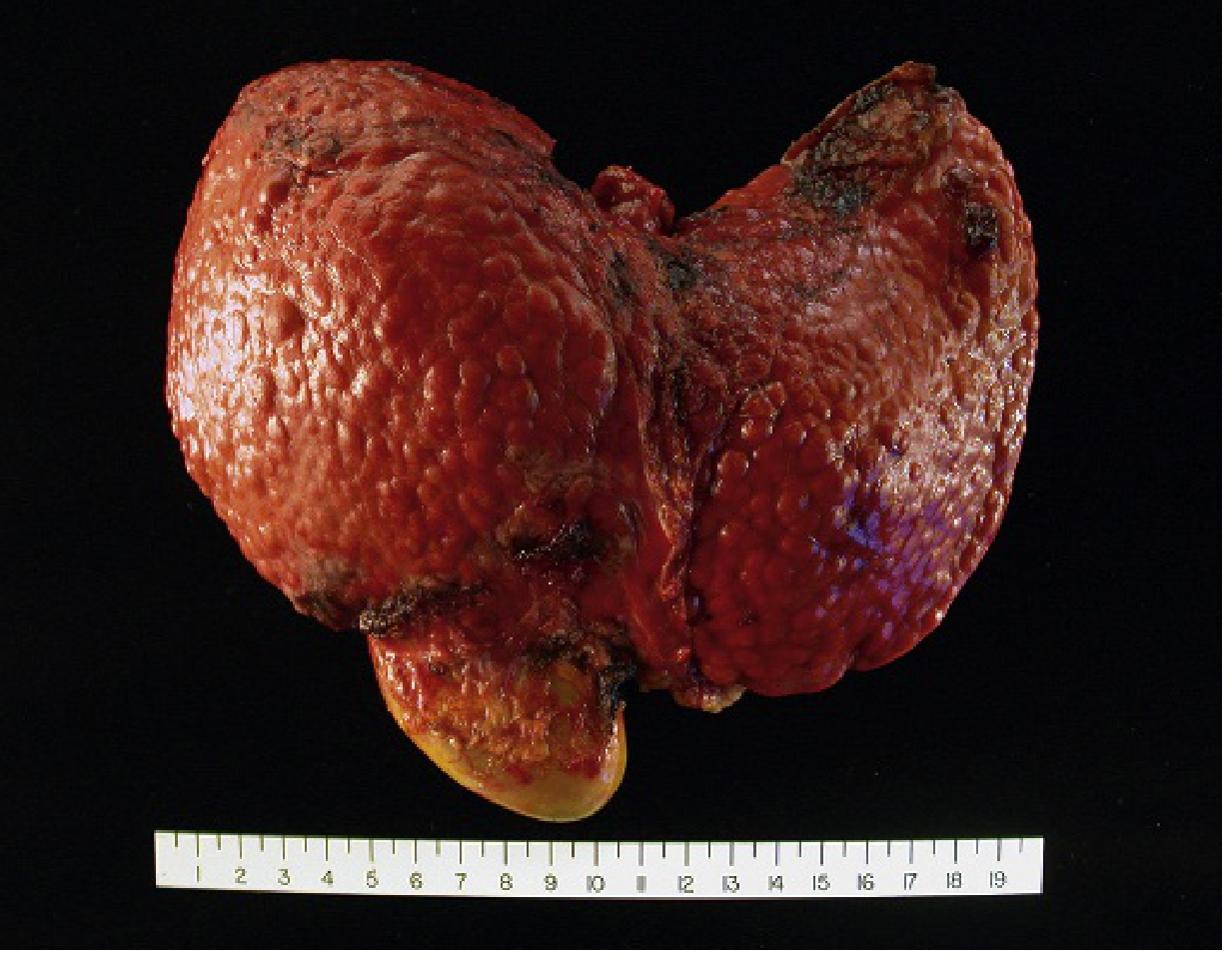
Gross specimen: Cirrhosis from chronic viral hepatitis — broad fibrous bands separate bulging regenerative nodules.
Etiology
| Category | Causes |
|---|---|
| Major (>90%) | Alcoholic liver disease, Non-alcoholic fatty liver disease (NAFLD/MASLD), Chronic hepatitis B |
| Other | Chronic hepatitis C, Primary biliary cholangitis, Primary sclerosing cholangitis, Autoimmune hepatitis |
| Metabolic | Hemochromatosis, Wilson disease, α1-Antitrypsin deficiency, Glycogen storage diseases |
| Vascular | Budd-Chiari syndrome, Right-sided heart failure, Veno-occlusive disease |
| Cryptogenic | No identifiable cause |
Goldman-Cecil Medicine, Table 139-1
Pathogenesis
Hepatic Stellate Cell Activation (Central Mechanism)
Hepatic stellate cells (Ito cells / perisinusoidal cells) reside in the space of Disse between hepatocytes and sinusoidal endothelial cells. Normally quiescent, serving as the main storage site for retinoids (Vitamin A).
In response to injury, stellate cells:
- Lose their vitamin A stores
- Proliferate and develop a prominent rough ER
- Secrete extracellular matrix — collagen types I and III, sulfated proteoglycans, glycoproteins
- Transform into contractile myofibroblasts
Collagen deposition in the space of Disse causes "capillarization" of sinusoids — the normally fenestrated endothelium loses its pores (defenestration), reducing exchange between plasma and hepatocytes and narrowing the sinusoidal lumen.
Goldman-Cecil Medicine, p. 1615
Reversibility
Fibrosis — and even established cirrhosis — can partially regress with disease remission or cure. Scars become thinner and fragment; nodules coalesce into larger parenchymal islands. All cirrhotic livers show elements of both progression and regression simultaneously.
Complications
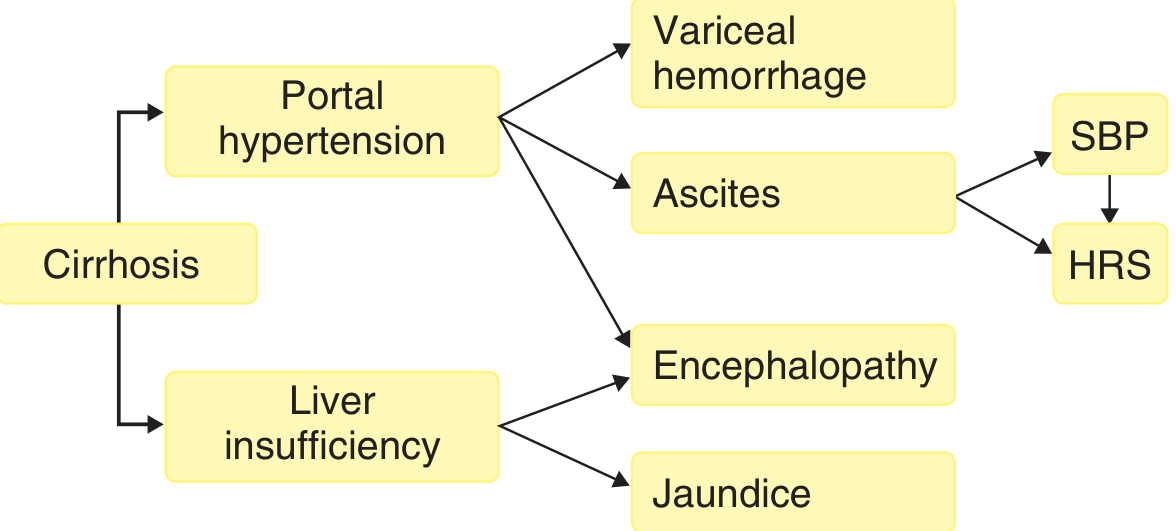
The two master pathways are portal hypertension and liver insufficiency.
1. Portal Hypertension & Hyperdynamic Circulation
Caused by:
- Fixed component: Fibrous tissue deposition + compression by regenerative nodules → ↑ sinusoidal vascular resistance
- Functional component: Deficiency of intrahepatic nitric oxide + enhanced vasoconstrictors → active vasoconstriction
Consequences:
- Splenomegaly → hypersplenism (thrombocytopenia, leukopenia)
- Portosystemic collaterals (coronary vein reversal → esophageal varices) — insufficient to decompress the portal system
- Splanchnic vasodilation (via ↑ NO, prostacyclin, endocannabinoids) → maintains portal hypertension
- Systemic arterial vasodilation → hyperdynamic circulation with ↑ cardiac output, ↓ SVR
2. Ascites
Arises from sinusoidal portal hypertension combined with hypoalbuminemia and secondary hyperaldosteronism. Managed with:
- Sodium restriction (88 mmol/day)
- Diuretics: spironolactone (first-line) ± furosemide
- Large-volume paracentesis + IV albumin for refractory/tense ascites
- TIPS (transjugular intrahepatic portosystemic shunt) for refractory cases
3. Spontaneous Bacterial Peritonitis (SBP)
~1/3 of hospitalized cirrhotics have bacterial infection; SBP is most common. Classic presentation: fever, abdominal pain, jaundice — but up to 1/3 initially present without abdominal symptoms (encephalopathy, AKI, or shock instead).
Diagnosis: Ascitic fluid PMN ≥ 250 cells/mm³. Treatment: cefotaxime (or similar 3rd-gen cephalosporin) + IV albumin.
4. Hepatic Encephalopathy (HE)
Occurs at ~2–3% per year. A spectrum from subclinical to coma:
| Grade | Features |
|---|---|
| 1 | Sleep-wake inversion, forgetfulness |
| 2 | Confusion, bizarre behavior, disorientation |
| 3 | Lethargy, profound disorientation |
| 4 | Coma |
Hallmark physical finding: asterixis (flapping tremor). Characteristic breath: fetor hepaticus. Mechanism: portosystemic shunting + liver insufficiency → ammonia accumulation + systemic inflammation.
Treatment: Lactulose (↓ ammonia production), rifaximin (second-line/prevention), treat precipitants.
5. Hepatorenal Syndrome (HRS)
Functional prerenal AKI in cirrhosis with ascites. Mechanism: maximal splanchnic vasodilation → renal artery vasoconstriction.
| Type | Characteristics |
|---|---|
| HRS-AKI (Type 1) | Rapidly progressive — creatinine doubles within 2 weeks; poor prognosis |
| HRS-non-AKI (Type 2) | Slowly progressive; associated with diuretic-refractory ascites |
Treatment: Vasoconstrictors (terlipressin preferred; norepinephrine + midodrine/octreotide where unavailable) + IV albumin. Definitive: liver transplantation.
6. Variceal Hemorrhage
Most dangerous acute complication. 20–30% mortality per episode. Primary prophylaxis: non-selective beta-blockers (propranolol/nadolol) or endoscopic variceal ligation (EVL).
Acute management: Endoscopic variceal ligation + terlipressin/somatostatin analogues + prophylactic antibiotics (ceftriaxone).
7. Other Complications
- Pruritus from cholestasis (bile salt accumulation)
- Coagulopathy (↓ synthesis of clotting factors)
- Hepatopulmonary syndrome (intrapulmonary vascular dilation → hypoxemia)
- Portopulmonary hypertension
- Hepatocellular carcinoma (HCC) — surveillance with ultrasound ± AFP every 6 months
Staging & Prognostic Scores
Compensated vs. Decompensated
- Compensated: Asymptomatic; no ascites, bleeding, encephalopathy, or jaundice. Median survival >12 years.
- Decompensated: Any of the above complications present. Median survival 2 years.
Child-Turcotte-Pugh (CTP) Score
Assesses: bilirubin, albumin, PT/INR, ascites, encephalopathy.
- Child A (5–6): Well-compensated
- Child B (7–9): Significant functional compromise
- Child C (10–15): Decompensated, poor prognosis
MELD Score
MELD = [0.957 × ln(Cr) + 0.378 × ln(bili) + 1.12 × ln(INR) + 0.643] × 10
MELD-Na adjusts for serum sodium when initial MELD ≥ 12 — the primary tool for liver transplant organ allocation.
Management Principles
| Goal | Intervention |
|---|---|
| Treat underlying cause | Antivirals (HBV/HCV), alcohol cessation, weight loss (MASLD) |
| Prevent decompensation | Non-selective beta-blockers, surveillance endoscopy |
| Manage ascites | Na restriction, spironolactone ± furosemide, paracentesis + albumin |
| Prevent/treat SBP | Norfloxacin prophylaxis in high-risk; cefotaxime for acute SBP |
| Treat HE | Lactulose + rifaximin; correct precipitants |
| HCC surveillance | Ultrasound ± AFP q6 months |
| Definitive | Liver transplantation for end-stage disease (MELD ≥ 15 typically considered) |
Key Sources
- Robbins & Kumar Basic Pathology, pp. 597–599
- Goldman-Cecil Medicine, Chapter 139 (pp. 1614–1621)
This is a shared conversation. Sign in to Orris to start your own chat.