Lower motor neuron palsy what is it, its causes, pathophysiology, treatment and complications as well as presentation in detail?
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full, detailed answer.
Lower Motor Neuron (LMN) Palsy - Comprehensive Overview
1. Definition
A lower motor neuron (LMN) palsy is weakness or paralysis caused by damage to the motor neurons that directly innervate skeletal muscle - specifically the alpha motor neurons in the anterior horn of the spinal cord and their axons as they travel through the ventral roots, plexuses, and peripheral nerves to the neuromuscular junction.
The LMN is the final common pathway for all motor output. Every voluntary movement, reflex, and postural adjustment must ultimately pass through it. Adams and Victor define the motor unit as "the motor neuron, its axon, and the muscle fibers they innervate" - and LMN palsy results from disruption anywhere along this unit.
- Adams and Victor's Principles of Neurology, 12th Ed.
2. Anatomy of the Lower Motor Neuron
The LMN system comprises:
- Alpha motor neurons in the anterior (ventral) horn of the spinal cord, organized somatotopically:
- Medial neurons supply axial/trunk muscles
- Lateral neurons supply appendicular (limb) muscles
- Axons exiting via ventral spinal roots
- Spinal nerves (formed by union of ventral + dorsal roots)
- Plexuses (brachial, lumbosacral)
- Peripheral nerves to the neuromuscular junction
- Cranial nerve motor nuclei in the brainstem (CN III, IV, V, VI, VII, IX, X, XI, XII) for face and bulbar musculature
The motor unit includes the alpha motor neuron plus all muscle fibers it innervates (ranging from a handful in extraocular muscles to 1,000+ in large limb muscles). All force gradation is achieved by recruiting more motor units and varying their firing frequency.
- Adams and Victor's Principles of Neurology, 12th Ed., pp. 66-67
3. Causes
Hereditary/Genetic
| Condition | Gene/Mechanism |
|---|---|
| Spinal Muscular Atrophy (SMA) Types I-IV | SMN1 mutations (autosomal recessive) |
| Kennedy disease (X-linked spinobulbar muscular atrophy) | CAG repeat expansion in androgen receptor gene |
| Hereditary motor neuropathies (distal SMAs) | Multiple genes |
| GM2 gangliosidosis (hexosaminidase deficiency) | HEXA/HEXB mutations |
Infective
- Acute poliomyelitis - poliovirus destroys anterior horn cells; the classic LMN palsy cause historically
- West Nile virus encephalitis - can produce acute flaccid paralysis
- Enterovirus 71 and other enteroviruses
- Rabies virus
- HIV-associated motor neuron disorder
- Lyme disease (Borrelia burgdorferi)
Acquired/Degenerative
- Amyotrophic Lateral Sclerosis (ALS) - most common motor neuron disease; affects both UMN and LMN
- Post-polio syndrome - late-onset LMN weakness decades after acute polio
- Multifocal motor neuropathy (MMN) - immune-mediated, anti-GM1 antibodies
- Monomelic focal/segmental SMAs
Toxic
- Lead toxicity - classical "wrist drop" from radial motor neuropathy
- Mercury toxicity
- Post-irradiation motor neuron syndrome - delayed LMN syndrome after spinal/paraspinal radiation
- Neurolathyrism, Konzo (plant toxins in famine settings)
Structural/Compressive
- Disc herniation with root compression
- Foraminal stenosis
- Tumors compressing nerve roots or peripheral nerves
- Trauma to peripheral nerves or nerve roots
Immune/Inflammatory
- Guillain-Barré syndrome (acute inflammatory demyelinating polyneuropathy)
- Acute motor axonal neuropathy (AMAN) - axonal GBS variant
- Vasculitic neuropathy
- Multifocal motor neuropathy
Metabolic/Endocrine
-
Hyperthyroidism, hyperparathyroidism, hypoglycemia (can cause motor neuron overactivity or degeneration)
-
Copper deficiency myelopathy
-
Goldman-Cecil Medicine, International Ed.
4. Pathophysiology
The Motor Unit and Neurotransmission
Alpha motor neurons use acetylcholine at the neuromuscular junction. The descending corticospinal fibers use glutamate/aspartate as excitatory transmitters. Renshaw cells provide recurrent inhibition via glycine. When an alpha motor neuron fires, all muscle fibers in its motor unit contract simultaneously ("all-or-none" for the unit).
What Happens When LMNs Are Destroyed
When the LMN or its axon is damaged:
- Voluntary, postural, and reflex movements are abolished in that muscle
- Flaccidity develops immediately - the muscle becomes lax because it is cut off from both direct neural drive and reflex arcs. Normal resting muscle tone depends on the tonic activity of gamma motor neurons keeping spindles sensitized; destroying alpha neurons removes the output arm entirely.
- Hypotonia/Atonia - the muscle offers no resistance to passive stretch
- Muscle spindle reflexes are lost - the reflex arc is broken (afferent signal cannot be completed into a motor response), causing areflexia
- Denervation atrophy begins within days: the muscle is reduced to 20-30% of its original bulk within 3-4 months. Denervated muscle fibers undergo atrophy from loss of trophic factors (neurotrophins) normally supplied by the motor nerve
- Fibrillations appear on EMG within 1-2 weeks of denervation - these are spontaneous contractions of individual muscle fibers, NOT visible to the naked eye
- Fasciculations - spontaneous firing of an entire motor unit, visible as a brief muscle twitch under the skin; indicates irritation/dying anterior horn cells or axons
- Wallerian degeneration occurs in the distal axon distal to the site of injury
- Electrodiagnostic changes: reduced motor unit potentials, fibrillation potentials, positive sharp waves, fasciculation potentials
Distinction by Lesion Site
- Anterior horn cell disease (e.g., polio, SMA, ALS): pure LMN signs with NO sensory loss, since dorsal horn and sensory nerves are spared
- Ventral root lesion: pure motor loss in a root distribution
- Mixed peripheral nerve lesion: both motor AND sensory deficits in peripheral nerve distribution
- Neuromuscular junction disease (myasthenia gravis, Lambert-Eaton): fatigable weakness, normal reflexes initially - not strictly LMN palsy
The presence or absence of sensory changes is a critical localizing feature: "the combination of flaccid, areflexic paralysis with sensory changes usually indicates the involvement of mixed motor and sensory nerves or of both anterior and posterior roots. If sensory changes are absent, the lesion must be situated in the anterior gray matter of the spinal cord, in the anterior roots, or in a purely motor branch."
- Adams and Victor's Principles of Neurology, 12th Ed.
5. Clinical Presentation (Signs and Symptoms)
Cardinal Signs of LMN Lesion
| Sign | LMN Lesion | UMN Lesion (for comparison) |
|---|---|---|
| Weakness | Yes | Yes |
| Atrophy | Yes (pronounced) | No (mild disuse only) |
| Fasciculations | Yes | No |
| Reflexes | Decreased/Absent | Increased |
| Tone | Decreased (flaccid) | Increased (spastic) |
| Babinski sign | Absent | Present |
| Clonus | Absent | Present |
- Neuroanatomy through Clinical Cases, 3rd Ed., Table 6.4
Detailed Features
1. Flaccid Weakness / Paralysis
- The muscle cannot generate force - voluntary power is lost in proportion to how many motor units are destroyed
- Partial involvement gives paresis; complete involvement gives plegia
- Distribution follows anatomic patterns:
- Anterior horn lesion: segmental pattern (myotomal), no sensory loss
- Root lesion: dermatomal sensory loss + myotomal weakness
- Peripheral nerve lesion: weakness + sensory loss in that nerve's territory
- Plexus lesion: complex pattern spanning roots/nerves
2. Hypotonia / Flaccidity
- On examination: arm/leg feels "limp," no resistance to passive movement
- Loss of normal postural tone
- Limbs may adopt unusual positions due to gravity
3. Areflexia / Hyporeflexia
- Deep tendon reflexes (biceps, triceps, knee jerk, ankle jerk) are reduced or absent at the affected level
- Superficial reflexes also absent in that territory
- No clonus, no Babinski sign (plantar response remains flexor or absent)
4. Muscle Atrophy
- Visible wasting of the affected muscle group
- Develops within weeks of denervation; severe by 3-4 months
- Helps localize the level and duration of damage
5. Fasciculations
- Visible spontaneous twitches of muscle fascicles (motor units firing spontaneously)
- Strongly suggests anterior horn cell or proximal axon irritation/degeneration
- Common in ALS, polio, SMA; seen in benign fasciculation syndrome too
6. Muscle Cramps
- Painful involuntary contractions; common in ALS and early denervation
7. No Babinski / No Clonus / No Spasticity
- The absence of these upper motor neuron signs distinguishes LMN from UMN palsy
Distribution Patterns by Localization
| Site | Pattern of Weakness | Sensory Involvement |
|---|---|---|
| Anterior horn (e.g., polio) | Segmental/myotomal, asymmetric | None |
| Ventral root | Myotomal | None (or minimal) |
| Dorsal + ventral root | Myotomal + dermatomal | Yes |
| Peripheral nerve | Distal nerve distribution | Yes (mixed nerve) |
| Plexus | Multiple roots/nerves, one limb | Yes |
| Neuromuscular junction | Proximal > distal, fatigable | None |
Cranial Nerve LMN Palsy (Bulbar)
- Facial nerve (CN VII) LMN palsy - affects the ENTIRE ipsilateral face (upper and lower), including forehead wrinkling and eye closure (Bell's palsy). This distinguishes it from UMN facial palsy, which spares the forehead (bilateral cortical representation)
- Hypoglossal (CN XII) LMN palsy: tongue wasting, fasciculations, deviates TOWARD the side of the lesion
- Vagus/glossopharyngeal (CN X/IX) LMN lesion: hoarseness, dysphagia, palate deviates away from lesion
- Bulbar involvement with LMN features = bulbar palsy (LMN type)
6. Investigations
- Electromyography (EMG): fibrillation potentials, positive sharp waves, fasciculation potentials, reduced recruitment of motor units
- Nerve conduction studies (NCS): reduced compound motor action potential (CMAP) amplitude in axonal damage; slowed conduction velocity in demyelination
- MRI spine/brain: to identify structural causes (disc herniation, cord compression, anterior horn signal change in polio/ALS)
- Muscle biopsy: shows grouped atrophy (denervation pattern) vs. random fiber atrophy (myopathic)
- Serum CK: mildly elevated in denervation; significantly elevated in muscular dystrophies
- Genetic testing: SMN1 deletion for SMA; CAG repeat for Kennedy disease
- CSF analysis: elevated protein in GBS
- Anti-GM1 antibodies: for multifocal motor neuropathy
- Pulmonary function tests: FVC to monitor respiratory involvement (critical in ALS, SMA)
7. Treatment
Treatment is cause-specific. There is no universal cure for most LMN diseases, but significant disease-modifying and supportive options exist.
Disease-Modifying Treatments
ALS (the most common LMN/mixed motor neuron disease):
- Riluzole (antiglutamate agent, oral): modest survival benefit (~3 months prolongation), especially in bulbar-onset ALS. It opens SK channels and reduces excitotoxic glutamate signaling. - Adams and Victor, 12th Ed.
- Edaravone (free radical scavenger, IV/oral): slows functional decline in early-stage ALS in selected patients
- Tofersen (antisense oligonucleotide targeting SOD1): approved for SOD1-positive familial ALS; slows clinical decline
- Sodium phenylbutyrate + taurursodiol (AMX0035): reduces mitochondrial and ER stress; showed slower functional decline vs. placebo at 24 weeks (Paganoni et al.)
- Masitinib (tyrosine kinase inhibitor): under investigation
Spinal Muscular Atrophy:
- Nusinersen (antisense oligonucleotide, intrathecal): approved for all SMA types, increases SMN2 exon 7 inclusion to boost SMN protein
- Onasemnogene abeparvovec (gene therapy, IV): single-dose AAV9-SMN1 gene replacement; transformative in SMA type I infants
- Risdiplam (oral SMN2 splicing modifier): approved for all SMA types
Guillain-Barré Syndrome:
- IV immunoglobulin (IVIG) or plasmapheresis - equivalent efficacy; started early
- Supportive care: ventilatory support if FVC <20%, pain management, DVT prophylaxis
Multifocal Motor Neuropathy:
- IVIG - first line; must be repeated regularly
- Rituximab in refractory cases
Post-polio syndrome:
- No disease-modifying treatment; rehabilitation, energy conservation, orthotic devices
Bell's Palsy (CN VII LMN palsy):
- Prednisolone (oral steroids): reduce inflammation, improve recovery
- Acyclovir/Valacyclovir: added if HSV suspected (HSV-1 reactivation implicated in most cases)
- Eye protection (lubricant drops, tape eye shut) to prevent corneal exposure injury
Compressive/structural (disc herniation, tumor):
- Surgical decompression when appropriate
- Physical and occupational rehabilitation
Symptomatic/Supportive Management
- Respiratory support: non-invasive ventilation (BiPAP/NIV), mechanical ventilation in severe cases; FVC monitoring is essential
- Nutrition: PEG (percutaneous endoscopic gastrostomy) tube feeding when swallowing becomes unsafe; nutritional assessment
- Physical therapy: maintain strength in unaffected muscles, prevent contractures, joint protection
- Occupational therapy: adaptive devices, mobility aids, communication devices (AAC)
- Speech-language pathology: dysphagia management, communication aids
- Spasticity management: not applicable to pure LMN disease, but relevant in mixed (ALS)
- Pain management: muscle cramps treated with quinine, baclofen, or mexiletine; nociceptive pain with analgesics
- Psychological support and palliative care: quality of life, advanced directives
8. Complications
| Complication | Mechanism |
|---|---|
| Respiratory failure | Denervation of respiratory muscles (diaphragm via phrenic nerve, intercostals) in SMA, ALS, GBS; most common cause of death |
| Aspiration pneumonia | Bulbar palsy with dysphagia and loss of protective reflexes |
| Contractures | Denervated muscles fibrosed over time with joint immobility |
| Pressure sores / decubitus ulcers | Immobility in paralyzed limbs |
| DVT / pulmonary embolism | Immobility, muscle pump failure |
| Malnutrition / weight loss | Dysphagia, increased energy expenditure |
| Corneal exposure keratitis | CN VII (facial nerve) LMN palsy: inability to close eye (lagophthalmos) |
| Depression and anxiety | Psychological burden of progressive paralysis |
| Joint subluxation | Loss of periscapular or shoulder girdle tone (e.g., flail arm) |
| Urinary complications | Infrequent in pure LMN disease (bladder innervation often spared) |
| Scoliosis | Paraspinal muscle weakness in pediatric SMA |
| Death (respiratory/bulbar failure) | End-stage of progressive LMN disease (ALS median survival 2-5 years; SMA I death often <2 years without treatment) |
Summary: LMN vs UMN at a Glance
| Feature | LMN | UMN |
|---|---|---|
| Tone | Flaccid (decreased) | Spastic (increased) |
| Reflexes | Reduced/absent | Brisk/hyperreflexia |
| Babinski | Absent | Present |
| Atrophy | Prominent, early | Minimal (disuse) |
| Fasciculations | Present | Absent |
| Clonus | Absent | Present |
| Distribution | Focal/segmental/nerve | Hemi/para/quadriparesis |
| Sensation | Lost if mixed nerve involved | Lost in UMN pattern |
Sources:
- Adams and Victor's Principles of Neurology, 12th Edition - Chapters 3, 38
- Goldman-Cecil Medicine, International Edition - Chapter on LMN Disorders
- Neuroanatomy through Clinical Cases, 3rd Edition - Chapter 6 (Table 6.4, KCC 6.7)
- Ganong's Review of Medical Physiology, 26th Edition
Lower motor neuron palsy of face what is it, its causes, pathophysiology, treatment and complications as well as presentation in detail?
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, multi-source textbook content. Let me compile the full detailed answer.
Lower Motor Neuron Palsy of the Face - Complete Detailed Overview
What is LMN Facial Palsy?
LMN facial palsy is weakness or paralysis of all ipsilateral facial muscles caused by a lesion of the facial nerve (cranial nerve VII) at or below its nucleus in the pons - anywhere along the facial nerve's course from the pons to the facial muscles. This is in contrast to UMN facial palsy (e.g., from stroke), which spares the forehead because the upper face has bilateral cortical representation.
The hallmark: the entire half of the face is affected, including the forehead - the patient cannot wrinkle their forehead, close their eye, or move the lower face on the affected side.
Anatomy of the Facial Nerve (CN VII) - Critical for Localization
The facial nerve has a long, complex course through a narrow bony canal (the fallopian/facial canal) that makes it uniquely vulnerable to pressure-induced injury:
- Facial nucleus in the lower pons (pontine tegmentum) - the LMN cell bodies
- Nerve loops around the abducens (CN VI) nucleus, forming the facial colliculus
- Exits pons at the cerebellopontine angle (CPA), alongside CN VIII
- Enters internal auditory meatus (IAM)
- Meatal foramen (labyrinthine segment entry) - the narrowest point (0.68 mm) - the critical bottleneck in Bell's palsy
- Labyrinthine segment - gives off the greater petrosal nerve (parasympathetic to lacrimal gland) and a sensory branch to the external ear
- Geniculate ganglion (sensory ganglion, site of VZV latency in Ramsay Hunt syndrome)
- Tympanic segment - gives off the nerve to stapedius muscle
- Mastoid (vertical) segment - gives off the chorda tympani (taste to anterior 2/3 tongue, parasympathetic to submandibular/sublingual glands)
- Exits at stylomastoid foramen
- Passes through parotid gland, divides into temporal, zygomatic, buccal, marginal mandibular, and cervical branches
The level of the lesion determines which additional functions are lost beyond facial weakness. This is critical for clinical localization:
| Lesion Level | Facial Weakness | Lacrimation | Hyperacusis | Taste (ant. 2/3) |
|---|---|---|---|---|
| Pontine nucleus | Yes (+ CN VI palsy possible) | Normal | Normal | Normal |
| CPA/IAM | Yes | Abnormal (dry eye) | Abnormal | Abnormal |
| Above geniculate ganglion | Yes | Abnormal | Abnormal | Abnormal |
| Between geniculate & stapedius | Yes | Normal | Abnormal | Abnormal |
| Between stapedius & chorda tympani | Yes | Normal | Normal | Abnormal |
| Below chorda tympani (near stylomastoid foramen) | Yes | Normal | Normal | Normal |
| Peripheral branches (parotid/distal) | Partial only | Normal | Normal | Normal |
- Adams and Victor's Principles of Neurology, 12th Ed.; Neuroanatomy through Clinical Cases, 3rd Ed.
The Key Anatomical Distinction: LMN vs UMN Facial Palsy
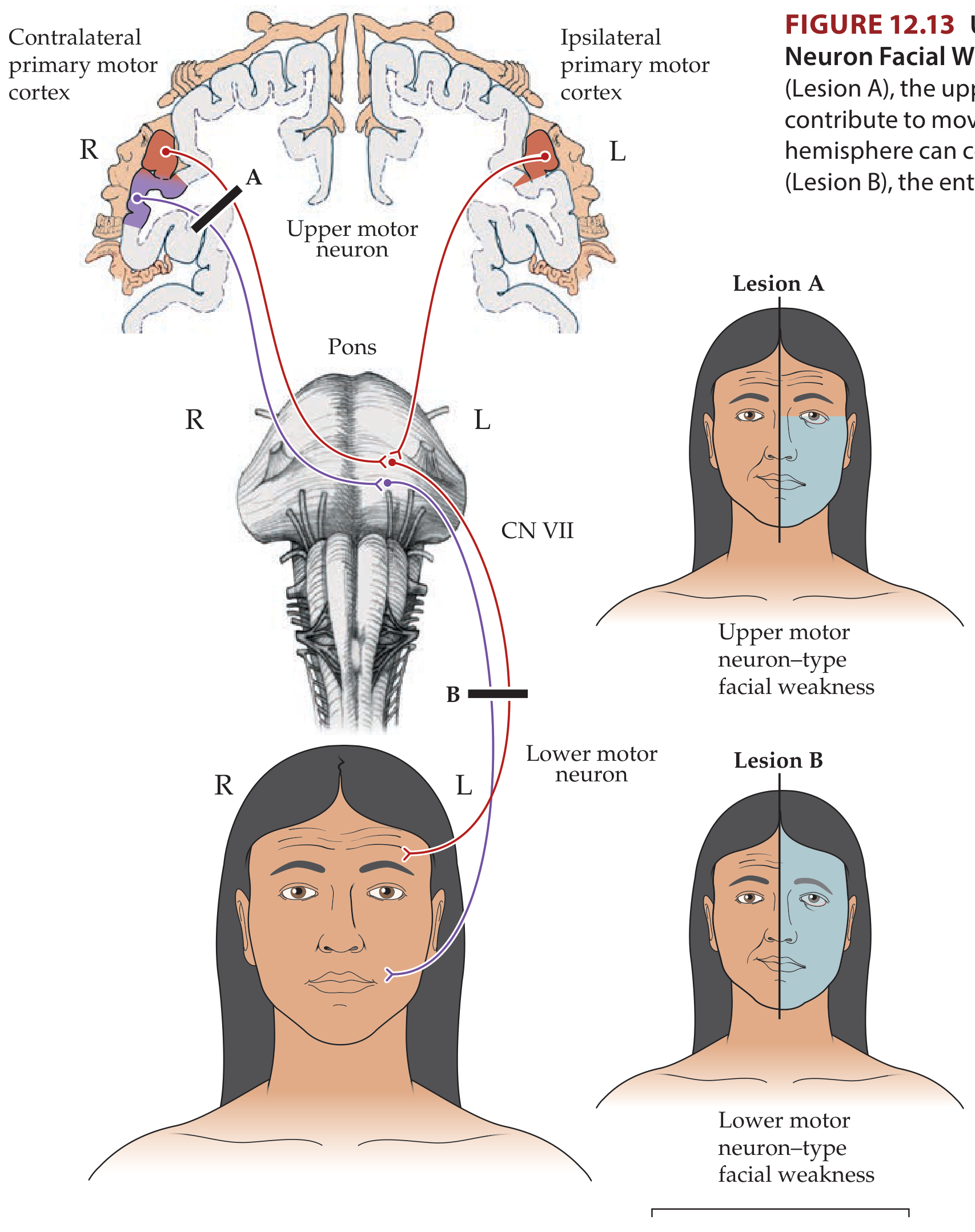
The upper face (forehead, orbicularis oculi superiorly) receives bilateral corticobulbar input from both cerebral hemispheres. Therefore, a unilateral UMN lesion (e.g., cortical stroke) leaves the forehead partially spared. In a LMN lesion, no cortical compensation is possible - the entire ipsilateral face is paralyzed.
-
LMN facial palsy: forehead involved, whole face paralyzed ipsilaterally
-
UMN facial palsy: forehead spared, lower face contralateral to lesion weakened
-
Neuroanatomy through Clinical Cases, 3rd Ed., KCC 12.3
Causes of LMN Facial Palsy
1. Idiopathic (Bell's Palsy) - Most Common (~70-75% of cases)
- Incidence 20-30 per 100,000 per year
- HSV-1 reactivation in the geniculate ganglion is the accepted mechanism - HSV-1 DNA found in endoneurial fluid and geniculate ganglion in studies using PCR
- Diagnosis of exclusion
2. Viral Infections
- Herpes zoster oticus (Ramsay Hunt syndrome) - VZV reactivation at the geniculate ganglion; 3-12% of facial palsies in adults; worse prognosis than Bell's palsy
- HIV infection - associated with facial nerve palsy (CSF pleocytosis typical)
- Epstein-Barr virus (infectious mononucleosis)
- Varicella (chickenpox) - may cause facial palsy 1-2 weeks after rash in children
- Poliomyelitis (historical)
3. Bacterial/Inflammatory
- Lyme disease (Borrelia burgdorferi) - one of the most common systemic causes, may be bilateral; associated with CSF pleocytosis
- Otitis media (acute or chronic) - bacterial spread along the dehiscent facial canal
- Malignant (necrotizing) external otitis - Pseudomonas aeruginosa in diabetics/immunocompromised
- Tuberculosis of the temporal bone (important in endemic areas)
- Leprosy - common in endemic areas, peripheral branch involvement
4. Structural/Compressive
- Acoustic neuroma (vestibular Schwannoma) - at the CPA/IAM; more often causes gradual involvement
- Cholesteatoma - erodes the facial canal
- Glomus jugulare tumor - jugular foramen
- Parotid gland tumors - invade extratemporal facial nerve
- Temporal bone fractures - longitudinal (perigeniculate) fractures in 20-25% of cases; transverse fractures more severe
- Carotid body tumors, dermoids, meningiomas
5. Granulomatous/Inflammatory Disease
- Sarcoidosis - facial palsy is common; may be bilateral; associated with Heerfordt syndrome (uveoparotid fever = parotid swelling + uveitis + facial palsy)
- Carcinomatous meningitis (leptomeningeal metastases)
6. Iatrogenic/Traumatic
- Birth trauma (forceps delivery) - commonest acquired cause in neonates; stylomastoid foramen region vulnerable
- Middle ear surgery/mastoidectomy
- Parotid surgery
- Penetrating head/neck trauma
7. Miscellaneous
-
Melkersson-Rosenthal syndrome - recurrent facial palsy + facial edema + scrotal tongue (autosomal dominant)
-
Guillain-Barré syndrome - bilateral facial palsy
-
Multiple sclerosis (demyelinating plaque in pons)
-
Möbius syndrome (congenital, bilateral facial diplegia due to absent facial nuclei)
-
Diabetes mellitus and hypertension - predisposing risk factors for Bell's palsy
-
Pregnancy (3rd trimester, especially with pre-eclampsia) - increased risk
-
Adams and Victor's Principles of Neurology, 12th Ed.; Goldman-Cecil Medicine; Shambaugh's Surgery of the Ear
Pathophysiology
Bell's Palsy (the Prototype)
The pathophysiology involves nerve swelling within the inelastic fallopian canal:
- Viral trigger: HSV-1 (which lies latent in the geniculate ganglion) reactivates, possibly triggered by stress, pregnancy, immunosuppression, or URI
- Inflammation and edema develop within the facial nerve
- Entrapment: The fallopian canal is rigid bone - the nerve cannot swell outward. The narrowest point, the meatal foramen (0.68 mm), acts as the critical bottleneck
- Conduction block (neuropraxia): edema prevents axoplasmic flow, blocking nerve impulse propagation. This is the mechanism in most cases - the axon itself remains intact
- If severe: axonal degeneration (Wallerian degeneration) begins distally - this leads to worse prognosis and prolonged recovery
- MRI gadolinium enhancement of the facial nerve at the geniculate ganglion region confirms the inflammatory reaction and correlates with severity
Degree of Nerve Injury
Using the Sunderland classification:
- Grade I (neuropraxia): conduction block only; full recovery expected within weeks
- Grade II (axonotmesis): axonal injury with intact endoneurium; recovery by axonal regrowth (~3 months); complete recovery likely
- Grade III-V (severe axonotmesis/neurotmesis): disruption of endoneurium or perineurium; aberrant regeneration, synkinesis, incomplete recovery
Ramsay Hunt Syndrome
VZV reactivates in the geniculate ganglion (where it lies latent after primary chickenpox). The virus then spreads anterograde along the facial nerve (causing palsy) and retrograde to skin supplied by the nervus intermedius (causing ear vesicles). The cochlear and vestibular branches of CN VIII are also frequently involved. The inflammatory reaction is more intense than in Bell's palsy, and "skip" areas of neuritis along the nerve explain why surgical decompression is not beneficial.
- Shambaugh's Surgery of the Ear; Adams and Victor's, 12th Ed.
Clinical Presentation
Core LMN Facial Palsy Signs
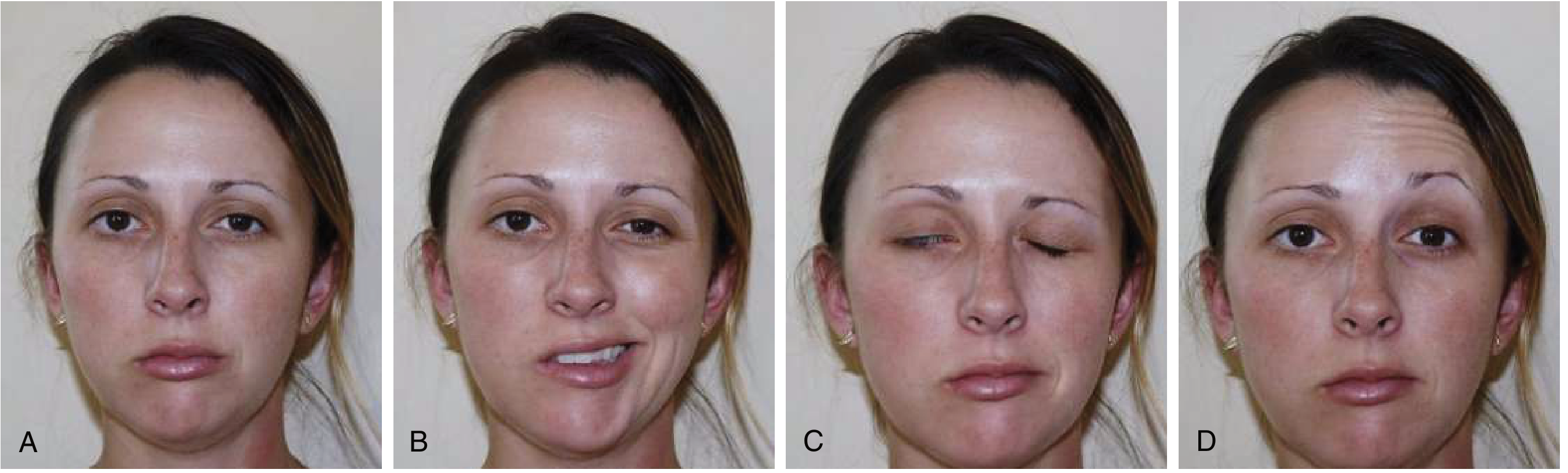
At rest:
- Facial asymmetry - affected side appears flat/expressionless
- Slight widening of the palpebral fissure (eye appears more open) due to loss of orbicularis oculi tone
- Loss of nasolabial fold on affected side
- Drooping of corner of mouth
On voluntary movement:
- Forehead - cannot wrinkle or raise eyebrow on affected side (differentiates LMN from UMN)
- Eye - cannot close eye (lagophthalmos); when attempting closure, eye rolls upward (Bell's phenomenon - a protective reflex); weak/absent blink reflex
- Nose - cannot flare nostril fully
- Mouth/Smile - mouth deviates to the unaffected side on smiling; cannot puff cheeks; food collects between cheek and gum (buccinator weakness); drooling
- Platysma - weak on affected side
Additional symptoms by lesion level:
| Symptom | Mechanism |
|---|---|
| Retroauricular/post-auricular pain | Inflammation at geniculate ganglion; involvement of somatic sensory fibers of CN VII |
| Hyperacusis (sounds painfully loud) | Stapedius muscle paralysis - cannot damp the ossicular chain |
| Loss of taste (anterior 2/3 of tongue) | Chorda tympani involvement |
| Dry eye (decreased lacrimation) | Greater petrosal nerve involvement (parasympathetic to lacrimal gland) - note: tearing may actually increase due to irritation |
| Decreased saliva | Chorda tympani parasympathetic fibers to submandibular/sublingual glands |
| Ear vesicles, otalgia | VZV reactivation = Ramsay Hunt syndrome |
| Sensorineural hearing loss + vertigo | CN VIII involvement (Ramsay Hunt) |
Bell's Palsy - Specific Time Course
- Onset: acute, often noticed in the mirror in the morning
- Progression: reaches maximum paralysis in ~48 hours in 50% of cases; practically all within 3-4 days
- Pain: retroauricular pain may precede palsy by 1-2 days (in about 50% of patients)
- Numbness: subjective facial fullness/numbness may occur; rarely objective hypoaesthesia
- Taste disturbance: present in most initially; usually resolves within 2 weeks
- Onset should NOT be slowly progressive over weeks - that suggests tumor
Ramsay Hunt Syndrome - Specific Presentation
- Severe otalgia (often the first symptom)
- Vesicles appearing in the external auditory canal, pinna, palate, tongue within 3-5 days of facial palsy onset
- Facial palsy (may be rapidly progressive and severe)
- Sensorineural hearing loss
- Vertigo, nausea, nystagmus
- CN V, IX, X, XI, XII may all be involved in severe cases
- VZV without skin vesicles = "zoster sine herpete" - can mimic Bell's palsy; PCR needed for diagnosis
Bilateral Facial Palsy
Causes to consider:
- Guillain-Barré syndrome (most common cause of simultaneous bilateral)
- Lyme disease (bilateral in ~25% of Lyme facial palsy)
- Sarcoidosis / Heerfordt syndrome
- HIV infection
- Infectious mononucleosis
- Möbius syndrome (congenital)
Grading - House-Brackmann Scale
| Grade | Description | Details |
|---|---|---|
| I | Normal | Symmetric function in all areas |
| II | Mild dysfunction | Slight weakness on close inspection; normal symmetry at rest |
| III | Moderate dysfunction | Obvious difference but not disfiguring; complete eye closure with effort |
| IV | Moderately severe | Obvious weakness; disfiguring asymmetry; incomplete eye closure |
| V | Severe dysfunction | Only barely perceptible movement; asymmetry at rest |
| VI | Total paralysis | No movement at all |
- Cummings Otolaryngology, Head and Neck Surgery
Investigations
- Clinical diagnosis (Bell's palsy): no investigations required if classic presentation
- MRI with gadolinium: enhancement of facial nerve at geniculate ganglion; worse prognosis with more pronounced enhancement; also identifies CPA tumors, MS plaques, brainstem lesions
- CT temporal bone: longitudinal vs transverse fractures; cholesteatoma; tumor erosion
- Electroneurography (ENOG/ENoG): measures CMAP amplitude; if >90% degeneration compared to normal side = poor prognosis indicator; used to guide surgical decisions in Bell's palsy
- EMG: fibrillation potentials appear 10+ days after denervation; absence of voluntary MUPs in complete palsy
- Audiogram + vestibular testing: if Ramsay Hunt suspected
- Lyme serology (ELISA + Western blot): if tick exposure, erythema migrans, or bilateral palsy
- HIV serology: risk factors present
- ACE level, chest X-ray: sarcoidosis
- CSF analysis: if Lyme, HIV, or carcinomatous meningitis suspected (pleocytosis in both Lyme and HIV facial palsy)
- PCR (VZV, HSV): from ear vesicle fluid; can detect VZV before vesicles appear in Ramsay Hunt
Treatment
Bell's Palsy
Corticosteroids (first-line - Grade A evidence):
- Prednisolone 1 mg/kg/day (60-80 mg/day orally) for 10 days, then taper
- Must be started within the first 2 weeks to be beneficial; ideally within 72 hours of onset
- Reduces edema within the fallopian canal, preventing axonal degeneration
- Improves recovery rate from ~63% to ~83% at 3 months
- Caution: monitor blood glucose (hyperglycemia risk), blood pressure; add PPI/H2 blocker for GI protection
Antivirals (adjunct, controversial):
- Valacyclovir 500 mg TID (or acyclovir 400 mg 5x/day) for 7 days combined with steroids
- Used because of HSV-1 etiology; some trials show additive benefit over steroids alone in complete palsy; others show no benefit
- Given the low side-effect profile, most centers add antivirals for moderate-to-severe palsy
Eye protection (mandatory):
- Artificial tear drops (lubricant drops) during the day
- Eye ointment at night + taping the eye shut during sleep
- Eye patch/moisture chamber
- Referral to ophthalmology if corneal sensation reduced
Surgical decompression:
- Middle cranial fossa (MCF) approach to decompress the meatal foramen, labyrinthine segment, and geniculate ganglion
- Considered only when: (1) ENOG shows ≥90% degeneration AND (2) no voluntary EMG activity AND (3) onset within 2 weeks AND (4) patient <65 years
- Transmastoid decompression does not adequately decompress the critical meatal foramen - not recommended for Bell's palsy
- Evidence for decompression remains limited and controversial
Ramsay Hunt Syndrome
- Antiviral + corticosteroid combination is superior to corticosteroids alone
- Acyclovir 800 mg 5x/day for 7-10 days (or valacyclovir 1g TID for 7 days) + prednisolone
- Treatment duration 3 weeks (longer than Bell's palsy due to prolonged active phase)
- Treat bacterial superinfection of vesicles with oral antibiotics
- Surgical decompression NOT recommended (skip regions of neuritis, diffuse involvement)
Lyme Disease Facial Palsy
- Doxycycline 100 mg BD for 14-21 days (oral, if no CNS involvement)
- IV ceftriaxone if CNS involvement (CSF pleocytosis, meningitis)
Traumatic Facial Palsy
- Immediate (complete) palsy after temporal bone fracture: surgical exploration recommended
- Delayed/progressive palsy after trauma: observe, steroids; surgical repair if ENOG shows ≥90% degeneration with no voluntary EMG activity
- Longitudinal fractures (90% of temporal bone fractures): palsy in 20-25%, usually perigeniculate; generally better prognosis
- Transverse fractures: palsy in 50%, more severe, involves labyrinthine segment
Otitis Media Related Palsy
- IV antibiotics + ventilation tube insertion (myringotomy)
- Reserve mastoidectomy for non-responders
- Recovery usually good (~80% recover well)
Tumor-Related
- Treatment directed at the underlying tumor
- Facial nerve grafting if nerve divided during resection
Prognosis
- Bell's palsy: 70% recover completely within 1-2 months; 85% achieve near-normal function
- Good prognostic signs: early return of taste (within 1 week); some motor recovery in first 5-7 days
- Poor prognostic signs: complete paralysis; elderly age; >90% degeneration on ENOG; prominent enhancement on MRI
- ~15% have significant residual deformity; ~15% have asymmetric movement/synkinesis
- Recurrence in ~8% (both sides can be affected across episodes); raises suspicion for alternative etiology
- Ramsay Hunt: nearly half have significant residual dysfunction; poorer prognosis than Bell's palsy
Complications
| Complication | Mechanism | Notes |
|---|---|---|
| Exposure keratitis / corneal ulceration | Lagophthalmos (inability to close eye) causing corneal desiccation | Most serious acute complication; can cause blindness |
| Corneal perforation / scarring | Severe keratitis if untreated | Preventable with eye care |
| Synkinesis | Aberrant regeneration - facial nerve axons regrow to wrong muscles | Involuntary facial movements accompany voluntary ones (e.g., eye closing when smiling) |
| "Crocodile tears" (Bogorad syndrome) | Aberrant regeneration - salivary fibers grow along lacrimal fibers | Tearing during eating/salivation |
| "Jaw winking" | Aberrant regeneration - eye closes when jaw moves | |
| Hemifacial spasm | Aberrant regeneration + ephaptic transmission | Involuntary unilateral facial contractions; late sequela |
| Facial contracture | Chronic denervation + fibrosis | Paradoxical tightening of face on affected side |
| Facial asymmetry / cosmetic deformity | Incomplete recovery | Psychosocial impact, depression |
| Post-herpetic neuralgia | VZV nerve damage (Ramsay Hunt) | Chronic neuropathic pain in ear/face |
| Hearing loss | CN VIII involvement (Ramsay Hunt) | Can be permanent sensorineural loss |
| Vertigo/balance problems | Vestibular involvement (Ramsay Hunt) | |
| Dry eye | Lacrimal gland denervation | Chronic ocular surface damage |
Summary Comparison: LMN vs UMN Facial Palsy
| Feature | LMN Facial Palsy | UMN Facial Palsy |
|---|---|---|
| Forehead involved | Yes - cannot raise brow | No - spared (bilateral cortical supply) |
| Side affected | Ipsilateral to lesion | Contralateral to lesion |
| Eye closure | Impaired (Bell's phenomenon seen) | Usually preserved |
| Taste/lacrimation | May be affected (depending on level) | Normal |
| Hyperacusis | May be present | Absent |
| Associated signs | Ear vesicles (Ramsay Hunt), parotid mass | Hemiplegia, aphasia, dysphagia (UMN signs) |
| Common cause | Bell's palsy (HSV-1) | Stroke, tumor |
Sources:
- Adams and Victor's Principles of Neurology, 12th Edition - Chapter on Facial Nerve
- Goldman-Cecil Medicine, International Edition - Bell Palsy Chapter
- Shambaugh's Surgery of the Ear - Idiopathic Facial Paralysis
- Cummings Otolaryngology Head and Neck Surgery - Facial Nerve Chapters
- Neuroanatomy through Clinical Cases, 3rd Edition - KCC 12.3
This is a shared conversation. Sign in to Orris to start your own chat.