Lab investigations of myocardial infarction
Reading File
Finding Sources
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "biomarkers"[MeSH Terms] AND "diagnosis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I now have rich content from multiple authoritative textbooks. Let me compose the comprehensive answer.
Laboratory Investigations of Myocardial Infarction
Diagnostic Framework
The diagnosis of acute MI requires a rise and/or fall of cardiac troponin (cTn) values with at least one value above the 99th percentile upper reference limit (URL), combined with at least one of:
- Symptoms of myocardial ischemia
- New ischemic ECG changes
- Development of pathologic Q waves
- Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality
- Identification of a coronary thrombus by angiography or autopsy (Type 1 MI)
Symptom to Diagnosis: An Evidence-Based Guide, 4th Ed. | Harrison's Principles of Internal Medicine, 22nd Ed.
1. Cardiac Troponins (Primary Biomarker)
Troponins are the gold-standard and preferred biomarkers for diagnosing myocardial injury and MI.
Biochemistry
The troponin complex consists of three regulatory proteins:
- Troponin C - calcium-binding subunit (not cardiac-specific - same isoform as slow-twitch skeletal muscle)
- Troponin I (cTnI) - inhibitory subunit; cardiac-specific due to a unique 31-amino-acid N-terminal extension not present in skeletal muscle isoforms
- Troponin T (cTnT) - tropomyosin-binding subunit; cardiac-specific due to an 11-amino-acid amino-terminal residue
Both cTnI and cTnT are encoded by unique genes distinct from skeletal muscle isoforms, providing high cardiac specificity. Approximately 94-97% of troponin is bound within myofibrils; a smaller cytoplasmic fraction (3-6%) represents a more easily mobilizable pool.
Tietz Textbook of Laboratory Medicine, 7th Ed.
Release Mechanism
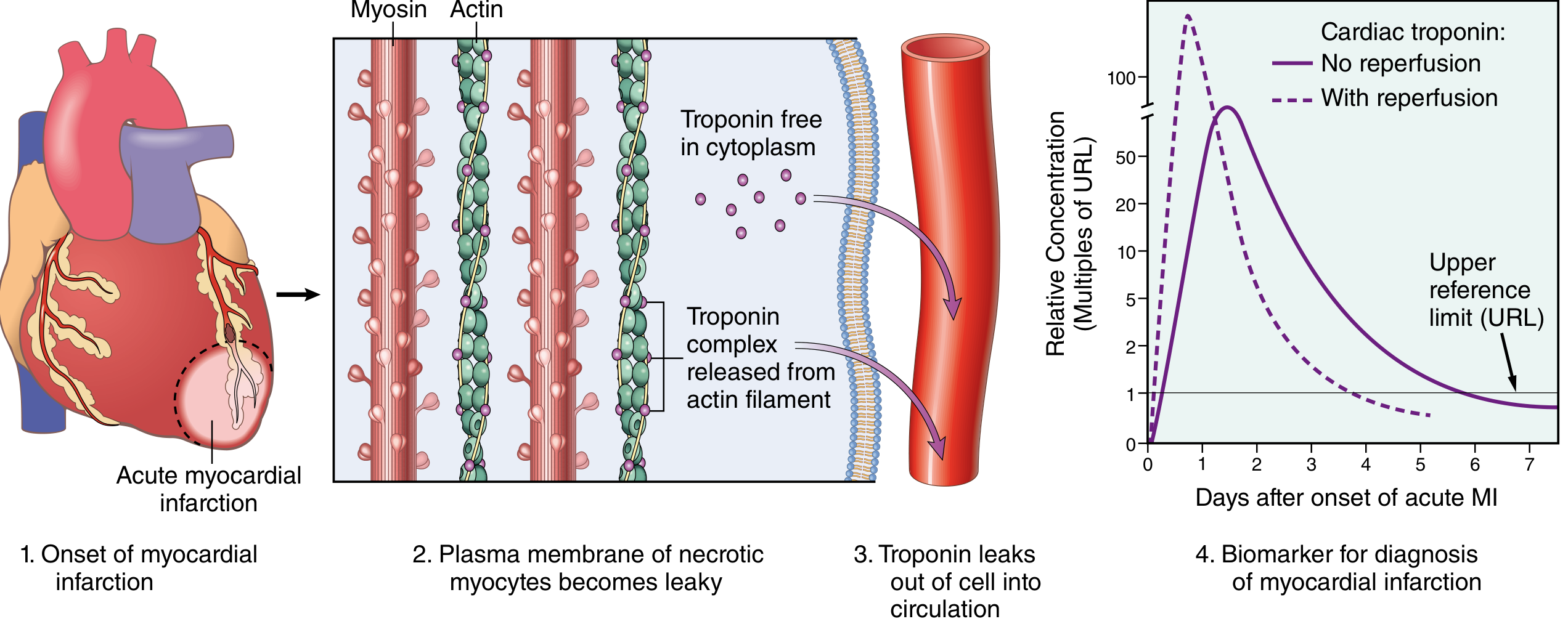
Fig: (1) Onset of MI. (2) Plasma membrane of necrotic myocytes becomes leaky. (3) Troponin leaks from cytoplasm and as released troponin-complex from actin filaments into the circulation. (4) Biomarker kinetics: without reperfusion (solid line) vs with reperfusion (dashed line - earlier, higher peak due to rapid washout). - Robbins, Cotran & Kumar
Kinetics
| Parameter | Cardiac Troponin (cTnI / cTnT) |
|---|---|
| Rises | 2-4 hours after onset |
| Peaks | 24-48 hours |
| Returns to normal | 7-10 days |
| Effect of reperfusion | Earlier, higher peak (rapid washout) |
- cTnI and cTnT are normally undetectable in serum
- Persist elevated for 7-10 days, allowing diagnosis even when presentation is delayed
- Serial measurements over 3-6 hours improve sensitivity for early presentations
Robbins & Kumar Basic Pathology | Robbins, Cotran & Kumar Pathologic Basis of Disease
Threshold
- Diagnosis requires at least one value above the 99th percentile URL of a healthy reference population
- Modern high-sensitivity cTn (hs-cTn) assays allow detection at much lower concentrations, enabling 0h/1h or 0h/2h rapid rule-out protocols
- Sex-specific URL values are recommended (women have lower 99th percentile thresholds)
Tietz Textbook of Laboratory Medicine, 7th Ed. | Harrison's Principles, 22nd Ed.
Causes of Troponin Elevation Other Than MI
Troponin can be elevated in other conditions - serial measurements help distinguish:
- Myocarditis, myocardial trauma
- Heart failure, pulmonary embolism
- Renal failure (reduced clearance), sepsis
- These causes do not follow the characteristic rise-and-fall pattern of acute MI
2. Creatine Kinase - MB Isoform (CK-MB)
CK-MB was historically the primary biomarker but has been largely replaced by troponin.
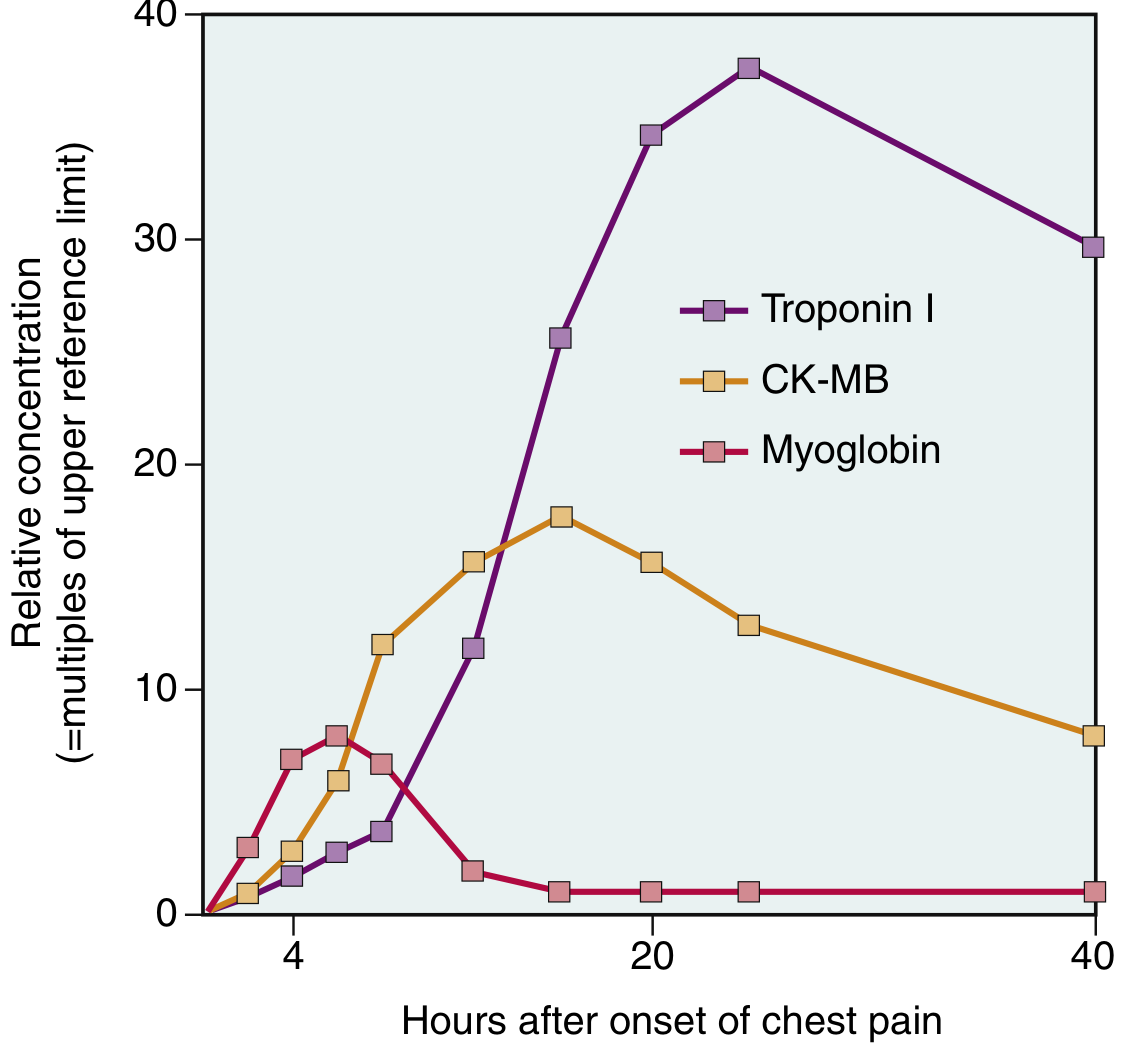
Fig: Comparative kinetics of Troponin I, CK-MB, and Myoglobin after chest pain onset (multiples of upper reference limit). Troponin I rises highest and persists longest. Myoglobin peaks earliest but is least specific. - Robbins & Kumar Basic Pathology
| Parameter | CK-MB |
|---|---|
| Rises | 2-4 hours |
| Peaks | 24-48 hours |
| Returns to normal | ~72 hours (3 days) |
| Specificity | Lower - present in both skeletal AND cardiac muscle |
Current role:
- No longer recommended as the primary marker for initial diagnosis of NSTEMI (Washington Manual of Medical Therapeutics)
- May still be used to detect reinfarction (since troponin remains elevated for 7-10 days, CK-MB's shorter window makes a second rise detectable)
- Occasionally used in perioperative settings (e.g., post-cardiac surgery)
Washington Manual of Medical Therapeutics | Robbins & Kumar Basic Pathology
3. Myoglobin
- A heme-containing oxygen-binding protein found in both cardiac and skeletal muscle
- Earliest to rise after MI onset (detectable within 1-3 hours)
- Returns to normal within 24 hours - shortest window
- Very low specificity - released by any muscle injury (skeletal, cardiac)
- Not recommended as a standalone diagnostic test but historically used as an early sensitive marker
- Useful as a negative predictor if levels are normal very early after symptom onset
4. Other Routine Lab Investigations
These are not diagnostic for MI but are obtained in all patients with suspected ACS:
| Test | Purpose |
|---|---|
| CBC (Complete Blood Count) | Detect anemia (worsens ischemia), leukocytosis (stress response), severe thrombocytopenia (alters antiplatelet therapy) |
| Basic Metabolic Panel / Electrolytes | Hypokalemia/hyperkalemia can mimic or worsen ECG changes; renal function affects drug dosing |
| Fasting Glucose / HbA1c | Stress hyperglycemia common; undiagnosed diabetes |
| Lipid Profile | Assess underlying atherosclerotic risk; guide statin therapy |
| Coagulation studies (PT, aPTT) | Baseline before anticoagulation |
| Renal function (Creatinine, GFR) | Affects contrast use (angiography), drug dosing |
| LDH (Lactate Dehydrogenase) | Historically used, peaks at 3-6 days, remains elevated up to 14 days - now largely replaced by troponin |
| AST (Aspartate Aminotransferase) | Historically used (peaks at 24-48h); non-specific, also elevated in liver disease |
Washington Manual of Medical Therapeutics
5. BNP / NT-proBNP (Natriuretic Peptides)
- BNP (B-type natriuretic peptide) is a biomarker of myocardial wall stress and ventricular dysfunction
- Elevated BNP in the setting of ACS indicates:
- Larger infarction size
- Worse outcomes / higher mortality
- Possible large infarction requiring urgent angiography (if severely elevated in a patient without known HF)
- Aids in distinguishing cardiac vs. pulmonary cause of dyspnea
Washington Manual of Medical Therapeutics | Tietz Textbook of Laboratory Medicine, 7th Ed.
6. Universal MI Classification and Biomarker Thresholds
The Fourth Universal Definition of MI (reflected in current guidelines) defines types based on pathophysiology, each with specific biomarker criteria:
| MI Type | Context | cTn Threshold |
|---|---|---|
| Type 1 | Spontaneous (plaque rupture/thrombosis) | >99th percentile URL with rise/fall |
| Type 2 | Supply/demand imbalance (e.g., spasm, anemia, tachycardia) | >99th percentile URL with rise/fall |
| Type 3 | Cardiac death before biomarkers available | Clinical/ECG criteria |
| Type 4a | PCI-related | >5x 99th percentile URL within 48h post-PCI |
| Type 5 | CABG-related | >10x 99th percentile URL within 48h post-CABG |
Harrison's Principles of Internal Medicine, 22nd Ed. | Sabiston Textbook of Surgery
Summary Table: Biomarker Kinetics
| Biomarker | Rises | Peaks | Normalizes | Specificity | Current Use |
|---|---|---|---|---|---|
| cTnI / cTnT | 2-4 h | 24-48 h | 7-10 days | Very high | First-line; gold standard |
| hs-cTn | 1-3 h | Earlier | 7-10 days | Very high | 0h/1h rapid rule-out |
| CK-MB | 2-4 h | 24-48 h | ~72 h | Moderate | Reinfarction detection |
| Myoglobin | 1-3 h | 6-9 h | ~24 h | Low | Largely abandoned |
| LDH | 24-48 h | 3-6 days | 8-14 days | Low | Historical only |
| AST | 8-12 h | 18-36 h | 3-4 days | Low | Historical only |
| BNP/NT-proBNP | Hours-days | Variable | Variable | Moderate | Risk stratification / HF |
Key Clinical Point: The laboratory diagnosis of MI is never made in isolation - it requires integration of biomarker kinetics (a rise and fall pattern), ECG findings, clinical symptoms, and imaging. A single elevated troponin without temporal change pattern may represent chronic myocardial injury (e.g., from renal failure or heart failure) rather than acute MI.
Robbins, Cotran & Kumar Pathologic Basis of Disease | Tietz Textbook of Laboratory Medicine, 7th Ed. | Symptom to Diagnosis: An Evidence-Based Guide, 4th Ed. | Washington Manual of Medical Therapeutics
Functional tumors of ovary
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive, well-sourced content. Let me compile the complete answer.
Functional Tumors of the Ovary
"Functional" ovarian tumors are those that secrete biologically active hormones (estrogens, androgens, or corticosteroids), producing distinct clinical syndromes. They belong primarily to the Sex Cord-Stromal Tumor category, which arises from the gonadal mesenchyme - the same embryonic tissue that gives rise to granulosa cells, theca cells, Sertoli cells, and Leydig cells.
Sex cord-stromal tumors account for 5-8% of all ovarian malignancies.
Classification
| Group | Tumor Types |
|---|---|
| Granulosa-Stromal Cell Tumors | Granulosa cell tumor (adult & juvenile), Thecoma, Fibroma |
| Androblastomas / Sertoli-Leydig Cell Tumors | Sertoli cell tumor, Sertoli-Leydig cell tumor, Leydig (hilus) cell tumor |
| Gynandroblastoma | Mixed male + female differentiation |
| Sex cord tumor with annular tubules | Associated with Peutz-Jeghers syndrome |
| Steroid Cell Tumors | Stromal luteoma, Leydig cell tumor, NOS |
Berek & Novak's Gynecology, Table 39-8
1. Granulosa Cell Tumors (GCT)
Overview
- Most common type of malignant sex cord-stromal tumor
- Account for 2-5% of all ovarian malignancies
- Considered low-grade malignancies
- Almost always unilateral (bilateral in only 2%)
- Tumors that are hormonally active show yellow coloration on cut surface (due to intracellular lipids)
Subtypes
| Feature | Adult GCT (95%) | Juvenile GCT (5%) |
|---|---|---|
| Age | Postmenopausal (two-thirds) | Under 30 years; may be prepubertal |
| Nuclei | Round/ovoid, pale, "coffee-bean" nuclei (longitudinal groove) | Rounder, more hyperchromatic nuclei |
| Mitoses | Few | Numerous |
| Driver mutation | FOXL2 (97%) | AKT1 (60%), GNAS (30%) |
| Follicle spaces | Call-Exner bodies | Large, irregular follicle spaces |
| Prognosis | Late recurrence (10-20 years later) | Mostly benign; malignant in ~10% |
Histology - Call-Exner Bodies (Pathognomonic)
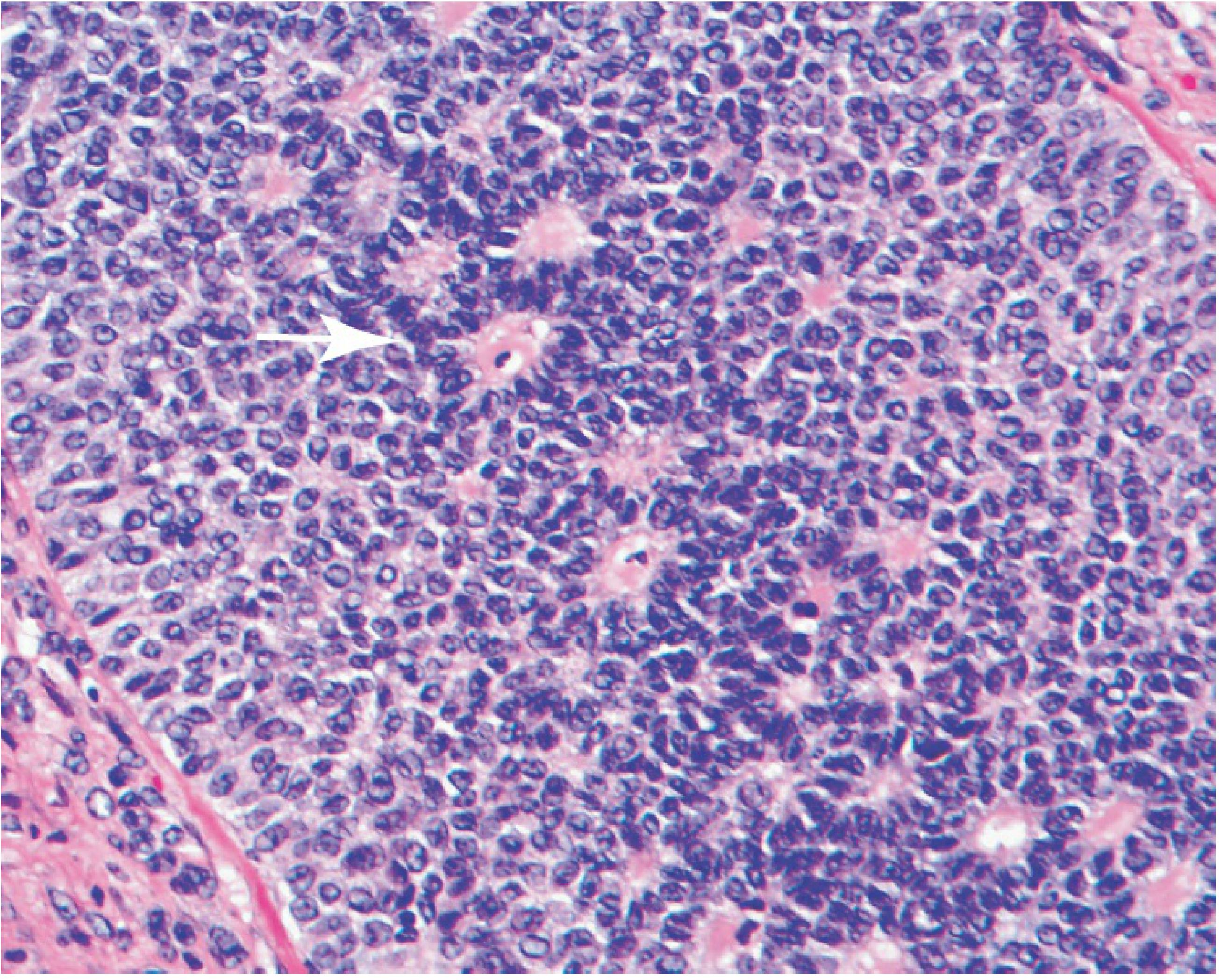
Granulosa cell tumor showing classic Call-Exner bodies (arrow) - small gland-like structures filled with acidophilic material recalling immature follicles (folliculoid pattern)
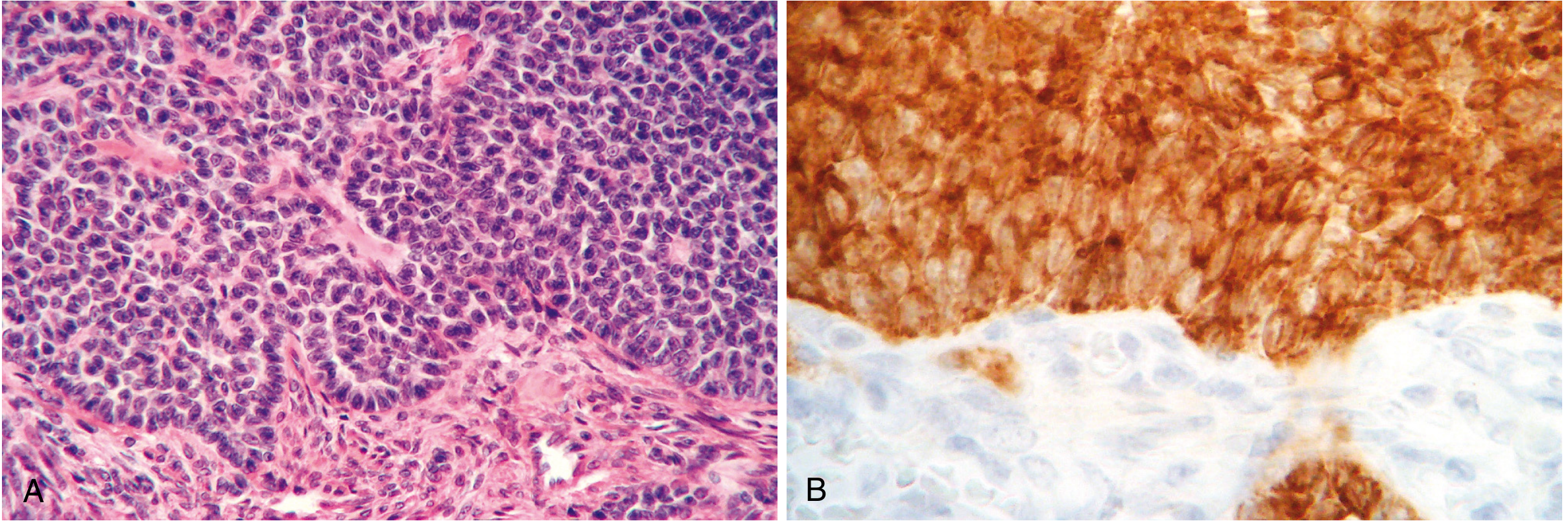
Fig. 22.41: (A) H&E showing tumor cells in sheets with small follicle-like Call-Exner bodies. (B) Strong immunohistochemical inhibin positivity - a hallmark of granulosa cell tumors
Hormone Secretion & Clinical Effects
About 70% of GCTs secrete estrogen (or androgens):
Estrogenic effects:
- Prepubertal girls: isosexual precocious puberty (breast development, premature menarche)
- Reproductive age women: menstrual irregularities, secondary amenorrhea, infertility
- Postmenopausal women: abnormal uterine bleeding (most common presenting symptom)
- Endometrial effects: cystic hyperplasia → endometrial hyperplasia → endometrial carcinoma in 10-15%; low-grade endometrial cancer co-exists in at least 5% of adult GCTs; endometrial hyperplasia in 25-50%
- Proliferative breast disease
Androgenic effects (occasional): masculinization/virilization of the patient
Tumor Markers
- Inhibin A and B - secreted by granulosa cells; inhibin B more frequently elevated. Useful for:
- Diagnosis (elevated inhibin in premenopausal woman with amenorrhea/infertility)
- Monitoring treatment response
- Detecting recurrence - lead time of ~12 months before clinical recurrence
- Anti-Müllerian Hormone (AMH) - elevated in 75% of adult GCTs preoperatively; >90% sensitivity for detecting recurrence
Molecular Genetics
- FOXL2 mutation (C134W) in 97% of adult GCTs - encodes a transcription factor important in granulosa cell development; nearly pathognomonic
- Molecular FOXL2 testing improves diagnostic accuracy (almost 30% of adult GCTs were misdiagnosed historically)
- FOXL2 mutation is absent in juvenile GCTs
Malignant Potential & Prognosis
- All GCTs are potentially malignant - impossible to predict biologic behavior from histology alone
- Malignant behavior (recurrence, extension): 5-25%
- Characteristically indolent course - recurrences may appear 10-20 years after original tumor removal
- 10-year survival: ~90-95% for Stage I
Robbins, Cotran & Kumar Pathologic Basis of Disease | Berek & Novak's Gynecology
2. Thecomas and Fibromas
Thecoma
- Composed of plump spindle cells with lipid droplets
- Represents theca cell differentiation
- Almost always benign (tumors composed predominantly of theca cells are almost never malignant)
- Hormonally active - typically estrogenic; produce endometrial hyperplasia, abnormal bleeding
- Usually unilateral, postmenopausal women
Fibroma
- Composed of well-differentiated fibroblasts with scant collagenous stroma
- Not usually hormonally active, but may occasionally be
- Unilateral in ~90% of cases; solid, hard, gray-white mass
- Account for ~4% of all ovarian tumors
- Meigs Syndrome - classic triad of:
- Ovarian fibroma (or thecoma)
- Ascites
- Right-sided pleural effusion
- (Resolves after tumor removal)
- Associated with Basal Cell Nevus Syndrome (Gorlin syndrome)
Robbins, Cotran & Kumar Pathologic Basis of Disease
3. Sertoli-Leydig Cell Tumors (Androblastoma)
Overview
- Recapitulate testicular architecture (Sertoli cells and Leydig cells)
- Extremely rare - <0.2% of ovarian cancers
- Peak incidence: 2nd and 3rd decades (75% under age 40)
- Unilateral; low-grade malignancies
- Associated with DICER1 gene somatic mutations (encodes endonuclease for micro-RNA processing)
Histology
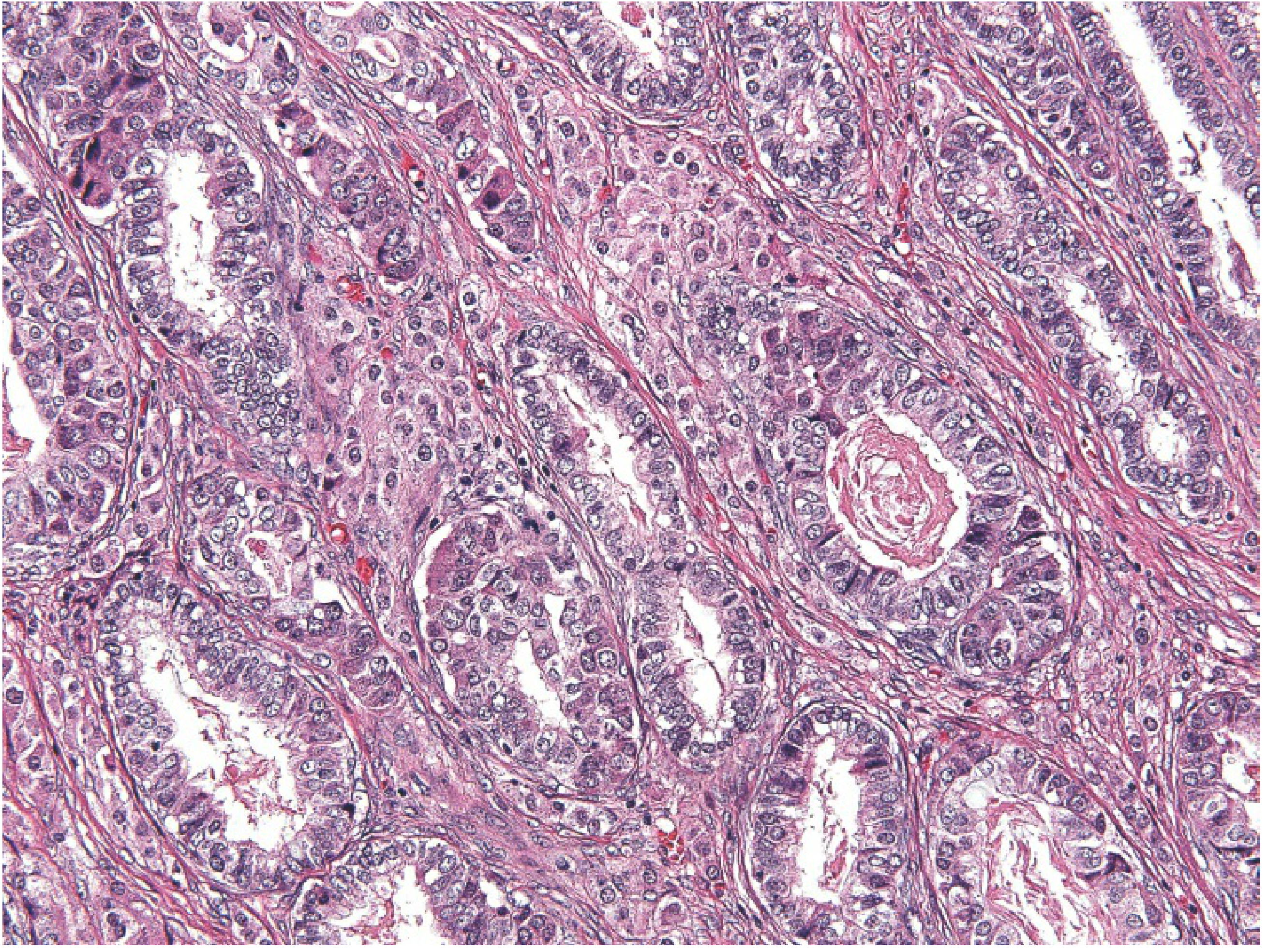
Sertoli-Leydig cell tumor: aggregates of eosinophilic Leydig cells in stroma adjacent to Sertoli cell tubules
Differentiation Grades
| Grade | Morphology |
|---|---|
| Well-differentiated | Tubules of Sertoli cells + Leydig cells interspersed with stroma |
| Intermediate | Immature tubules + large eosinophilic Leydig cells |
| Poorly differentiated (sarcomatoid) | Disorderly epithelial cell cords; Leydig cells may be absent |
| With heterologous elements | Mucinous glands, bone, or cartilage present |
Hormone Secretion & Clinical Effects
- Predominantly androgenic (testosterone and androstenedione elevated)
- Virilization/defeminization in 70-85% of patients:
- Oligomenorrhea → amenorrhea
- Breast atrophy
- Acne, hirsutism
- Clitoromegaly
- Voice deepening
- Receding hairline
- Rarely - estrogenic effects (isosexual precocity or postmenopausal bleeding)
- Plasma DHEAS normal or only slightly elevated (helps distinguish from adrenal source)
Prognosis
- Recurrence or metastasis: <5% (most are stage I, low grade)
- 5-year survival: 70-90%
- Most fatalities occur with poorly differentiated tumors
Berek & Novak's Gynecology | Robbins, Cotran & Kumar Pathologic Basis of Disease
4. Leydig Cell (Hilus Cell) Tumors
- Rare, unilateral tumors located in the ovarian hilus
- Composed of large lipid-laden Leydig cells with characteristic Reinke crystalloids (cytoplasmic crystalline inclusions - pathognomonic)
- Androgen-secreting → predominantly testosterone → mild-to-moderate virilization (hirsutism, voice changes, clitoromegaly)
- Milder virilization than Sertoli-Leydig cell tumors
- Almost always benign
- Treatment: surgical excision
5. Steroid Cell Tumors
- Account for <1% of ovarian neoplasms
- Most associated with virilization (most common)
- Some produce corticosteroids → obesity, hypertension, glucose intolerance (Cushing-like features)
- Rare cases of estrogen secretion and isosexual precocity
- ~20% may metastasize (especially if >8 cm)
- Treatment: surgical removal; no established role for radiation or chemotherapy
6. Gynandroblastoma and Sex Cord Tumor with Annular Tubules
- Gynandroblastoma: rare; contains both well-differentiated male (Sertoli/Leydig) and female (granulosa/theca) elements; may produce mixed hormonal effects
- Sex cord tumor with annular tubules (SCTAT): distinctive tumor with ring-shaped tubules; strongly associated with Peutz-Jeghers syndrome; may produce progesterone; occasionally malignant
Summary Table
| Tumor | Hormone Secreted | Main Clinical Effect | Key Feature | Malignant Potential |
|---|---|---|---|---|
| Granulosa cell tumor | Estrogen (mainly) | Precocious puberty, endometrial hyperplasia/cancer, AUB | Call-Exner bodies, FOXL2 mutation | Low-grade malignant; late recurrence |
| Thecoma | Estrogen | Endometrial hyperplasia, AUB | Lipid-laden spindle cells | Almost always benign |
| Fibroma | None (usually) | Meigs syndrome | Hard, gray-white mass | Benign |
| Sertoli-Leydig cell tumor | Androgen | Virilization, defeminization | Sertoli tubules + Leydig cells, DICER1 mutation | Low-grade malignant (<5% recur) |
| Leydig (hilus) cell tumor | Testosterone | Mild virilization | Reinke crystalloids | Benign |
| Steroid cell tumor | Androgens/corticosteroids | Virilization, Cushing-like | Lipid-rich cells | 20% malignant if >8 cm |
Robbins, Cotran & Kumar Pathologic Basis of Disease | Berek & Novak's Gynecology | Robbins & Kumar Basic Pathology | Guyton & Hall Textbook of Medical Physiology
Hemorrhagic disorders, classification and lab diagnosis for 20 marks
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a comprehensive 20-mark answer. Let me compile it.
Hemorrhagic Disorders: Classification and Laboratory Diagnosis
Introduction
Hemorrhagic disorders (hemorrhagic diatheses) are conditions in which bleeding occurs excessively, either spontaneously or following minor trauma. Normal hemostasis depends on three interacting components: (1) the vessel wall, (2) platelets, and (3) the coagulation cascade. A defect in any one - or combination - of these components leads to a bleeding disorder.
Classification
Hemorrhagic disorders are broadly classified based on the primary underlying defect:
HEMORRHAGIC DISORDERS
├── I. Vascular (Vessel Wall) Disorders
├── II. Platelet Disorders
│ ├── A. Quantitative (Thrombocytopenia)
│ └── B. Qualitative (Thrombocytopathy)
├── III. Coagulation Factor Disorders
│ ├── A. Hereditary
│ └── B. Acquired
└── IV. Mixed / Combined Defects (e.g., DIC, liver disease)
I. Vascular (Vessel Wall) Disorders
These are relatively common but rarely cause serious bleeding. Presentation is typically petechiae and purpura in skin/mucous membranes. Platelet count and coagulation tests (PT, PTT) are usually normal.
A. Hereditary
| Condition | Mechanism | Features |
|---|---|---|
| Hereditary Hemorrhagic Telangiectasia (Rendu-Osler-Weber) | Autosomal dominant; mutations in TGF-β signaling genes; dilated thin-walled vessels | Epistaxis, GI bleeding, mucous membrane telangiectasias; most serious vascular bleeding disorder |
| Ehlers-Danlos Syndrome | Collagen synthesis defect → weak vessel walls | Skin fragility, hypermobility, petechiae |
| Marfan Syndrome | Fibrillin defect | Aortic aneurysm, less prominent mucosal bleeding |
B. Acquired
| Condition | Mechanism |
|---|---|
| Infections (meningococcemia, septicemia, rickettsiae) | Vasculitis, endothelial damage, secondary DIC |
| Henoch-Schönlein Purpura (IgA vasculitis) | IgA immune complex deposition in vessel walls; purpura, abdominal pain, arthralgia, glomerulonephritis |
| Scurvy (Vitamin C deficiency) | Impaired collagen synthesis → weakened perivascular connective tissue |
| Cushing Syndrome / Corticosteroid use | Protein wasting → loss of perivascular extracellular matrix |
| Senile purpura | Age-related loss of perivascular support |
| Drug reactions | Immune complex deposition (leukocytoclastic vasculitis) |
| Perivascular amyloidosis | Weakening of vessel wall by amyloid deposits |
II. Platelet Disorders
A. Quantitative - Thrombocytopenia
Definition: Platelet count <150,000/μL. Spontaneous bleeding risk increases at <20,000/μL.
Bleeding pattern: Petechiae, mucocutaneous bleeding, epistaxis, gum bleeding - immediate after trauma.
Causes (Table 14.9 - Robbins):
1. Decreased Platelet Production
- Drug-induced: alcohol, thiazides, cytotoxic drugs
- Infections: measles, HIV
- Nutritional deficiency: B12, folate (megaloblastic states)
- Aplastic anemia
- Bone marrow replacement (leukemia, lymphoma, myeloma, metastases)
- Myelodysplastic syndrome
2. Increased Platelet Destruction
- Immune Thrombocytopenic Purpura (ITP) - most important:
- Autoantibodies (IgG) against platelet membrane glycoproteins (GpIIb/IIIa)
- Acute form: children, post-viral; Chronic form: adult women
- Megakaryocytes increased in bone marrow (compensatory)
- Lab: low platelet count, normal PT/PTT, normal bleeding time only if severe
- Thrombotic Thrombocytopenic Purpura (TTP):
- Deficiency of ADAMTS13 (metalloprotease that cleaves ultra-large vWF multimers)
- Pentad: thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, renal failure, neurologic changes
- Lab: fragmented RBCs (schistocytes) on peripheral smear, elevated LDH, low ADAMTS13 activity
- Hemolytic Uremic Syndrome (HUS): E. coli O157:H7 (Shiga toxin), complement dysregulation; mainly renal involvement
- Drug-induced immune thrombocytopenia: heparin (HIT), quinine
3. Sequestration
- Hypersplenism (any cause of splenomegaly)
4. Dilutional
- Massive blood transfusion with stored blood (low platelets)
B. Qualitative - Thrombocytopathy (Platelet Function Disorders)
Normal platelet count but abnormal platelet function; prolonged closure time (PFA-100) or historically prolonged bleeding time.
Hereditary:
| Disorder | Defect | Mechanism |
|---|---|---|
| Bernard-Soulier Syndrome | Deficiency of GpIb/IX | Impaired platelet adhesion to vWF and subendothelial matrix; autosomal recessive; giant platelets |
| Glanzmann Thrombasthenia | Deficiency of GpIIb/IIIa | Impaired platelet aggregation (no fibrinogen bridging between platelets); autosomal recessive; severe bleeding |
| Storage Pool Disorders | Deficient/abnormal dense granules (delta) or alpha granules | Defective release of ADP, thromboxane A2 → impaired platelet activation |
Acquired:
- Aspirin / NSAIDs: irreversible cyclooxygenase inhibition → impaired thromboxane A2 synthesis → impaired aggregation
- Uremia: complex defects in adhesion, secretion, and aggregation (requires dialysis or desmopressin for correction)
- Dysproteinemias (myeloma): paraproteins coat platelet surface
III. Coagulation Factor Disorders
Bleeding pattern: Deep tissue bleeding - hematomas, hemarthroses (joint bleeds), retroperitoneal bleeding - characteristically delayed after trauma (12-24 hours).
A. Hereditary Coagulation Factor Deficiencies
1. Hemophilia A (Factor VIII Deficiency)
- X-linked recessive; affects males; carrier females may occasionally bleed
- Accounts for 80% of hemophilias
- Severity based on factor VIII level:
- Severe: <1% activity → spontaneous bleeding into joints (hemarthrosis), muscles, CNS
- Moderate: 1-5% → bleeding with minor trauma
- Mild: 5-25% → bleeding with surgery/major trauma
- Lab: ↑PTT, Normal PT, Normal TT, Normal platelet count and bleeding time; specific factor VIII assay confirms
2. Hemophilia B (Christmas Disease - Factor IX Deficiency)
- X-linked recessive; clinically identical to Hemophilia A
- Lab: ↑PTT, Normal PT - identical pattern to Hemophilia A
- Distinguished only by specific factor assay
3. von Willebrand Disease (vWD) - Most common inherited bleeding disorder (~1% of population)
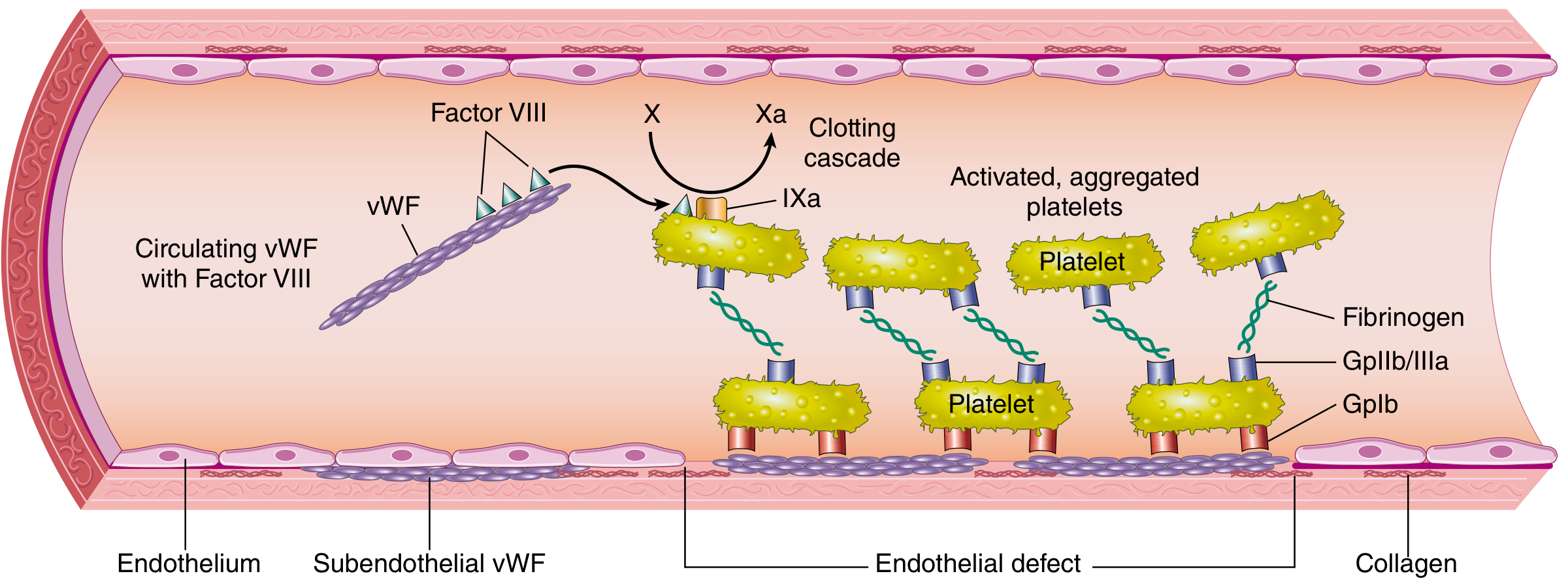
vWF circulates with Factor VIII, anchors platelets via GpIb to subendothelial collagen, and also bridges platelets via GpIIb/IIIa
- Autosomal dominant (Type 1, 2); autosomal recessive (Type 3)
- vWF defect impairs BOTH platelet adhesion AND factor VIII stability
| Type | Defect | vWF Antigen | vWF Activity | FVIII | Ristocetin |
|---|---|---|---|---|---|
| Type 1 (most common, 70-80%) | Quantitative ↓ (mild-moderate) | Low | Low | Low-normal | ↓ |
| Type 2 (qualitative) | Abnormal multimers (various subtypes) | Normal or low | Low | Variable | ↓ |
| Type 3 (most severe) | Near total absence | Very low/absent | Very low | Very low | Absent |
vWD Lab: ↑ PTT (due to ↓ Factor VIII), normal PT, ↓ vWF antigen, ↓ vWF activity (ristocetin cofactor), ↓ ristocetin-induced platelet agglutination (RIPA)
4. Other Rare Hereditary Factor Deficiencies (Goldman-Cecil Table 157-2):
| Factor Deficient | PT | aPTT | TT | Notes |
|---|---|---|---|---|
| Factor VII | ↑ | Normal | Normal | Extrinsic pathway only |
| Factor VIII (Hemophilia A) | Normal | ↑ | Normal | |
| Factor IX (Hemophilia B) | Normal | ↑ | Normal | |
| Factor XI | Normal | ↑ | Normal | Ashkenazi Jews |
| Factors V, X, Prothrombin | ↑ | ↑ | Normal | Both pathways |
| Fibrinogen (afibrinogenemia) | ↑↑ | ↑↑ | ↑↑ | All tests abnormal |
| Factor XIII | Normal | Normal | Normal | Requires specific assay; delayed wound healing |
| Vitamin K-dependent factors (II,VII,IX,X) | ↑↑ | ↑ | Normal | Warfarin / Vit K deficiency |
B. Acquired Coagulation Disorders
1. Vitamin K Deficiency
- Vitamin K required for carboxylation (activation) of factors II, VII, IX, X and anticoagulant proteins C and S
- Causes: malabsorption (biliary obstruction, celiac), newborns (hemorrhagic disease of newborn), warfarin therapy, prolonged antibiotic use
- Lab: ↑PT (first affected - Factor VII has shortest half-life), then ↑PTT; Normal TT; Normal platelet count
2. Liver Disease
- Liver synthesizes most clotting factors (I, II, V, VII, VIII partially, IX, X, XI)
- Results in multiple factor deficiencies + thrombocytopenia (portal hypertension-related splenomegaly) + ↓ clearance of activated factors
- Lab: ↑PT, ↑PTT, ↓fibrinogen, thrombocytopenia, ↑TT
3. Inhibitors / Autoantibodies
- Acquired inhibitors against specific factors (commonly Factor VIII) - rare
- Lab pattern mimics deficiency but does not correct on mixing study (1:1 mix of patient + normal plasma)
IV. Disseminated Intravascular Coagulation (DIC)
A paradigm of a combined/mixed hemorrhagic disorder - simultaneous thrombosis AND bleeding.
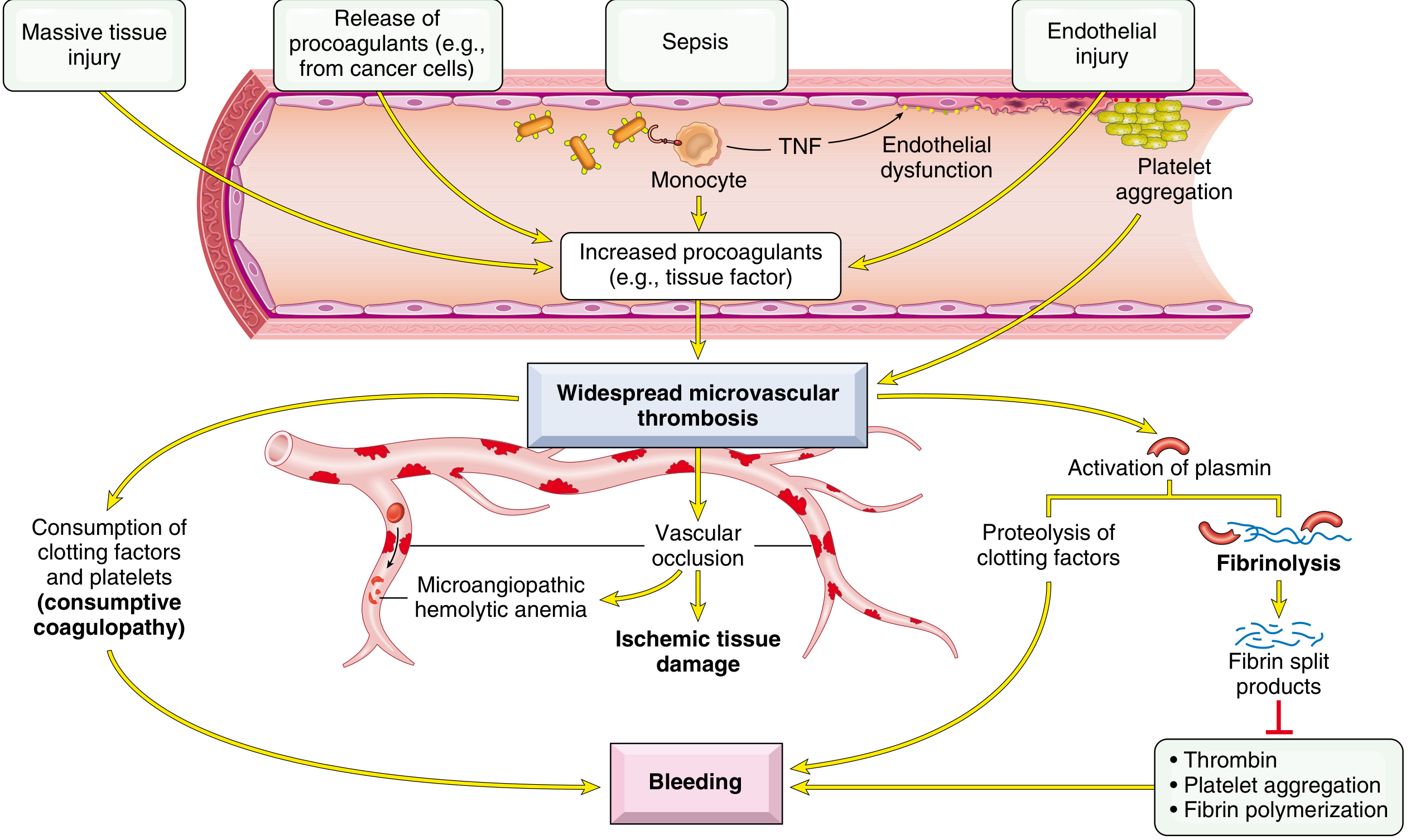
DIC pathophysiology: Triggers (sepsis, massive tissue injury, endothelial injury, cancer procoagulants) → widespread microvascular thrombosis → consumption of clotting factors and platelets (consumptive coagulopathy) + secondary fibrinolysis → bleeding
Pathophysiology
- Triggers → systemic activation of coagulation (via tissue factor release)
- Widespread fibrin deposition in microcirculation → organ ischemia + microangiopathic hemolytic anemia (RBC shearing through occluded vessels)
- Consumption of platelets and clotting factors → coagulopathy
- Secondary fibrinolysis activated → generates fibrin degradation products (FDPs/D-dimers) which further inhibit thrombin, platelet aggregation, and fibrin polymerization → hemorrhage
Common Triggers
- Sepsis (most common), Gram-negative endotoxemia
- Obstetric complications: amniotic fluid embolism, abruptio placentae, dead fetus syndrome
- Massive trauma, burns, extensive surgery
- Malignancy: acute promyelocytic leukemia (APL), adenocarcinomas (lung, pancreas, colon, stomach)
- Transfusion reactions, snake bites, Kasabach-Merritt syndrome
Lab Findings in DIC (Tintinalli Emergency Medicine Table 233-7)
| Test | Result | Comment |
|---|---|---|
| Platelet count | ↓ (most sensitive; progressive decline) | Consumption |
| Prothrombin time (PT) | ↑↑ (prolonged) | Factor consumption |
| aPTT | Usually ↑ | Factor consumption |
| Thrombin Time (TT) | ↑ | Low fibrinogen + FDPs inhibiting thrombin |
| Fibrinogen | ↓ (but may be initially normal - acute phase reactant) | <100 mg/dL = severe DIC |
| D-dimer | ↑↑ (most specific for fibrin breakdown) | Best marker for DIC activity |
| Fibrin Degradation Products (FDPs) | ↑↑ | Secondary fibrinolysis |
| Peripheral smear | Schistocytes (microangiopathic hemolytic anemia) | Red cell fragmentation in fibrin mesh |
| LDH | ↑ | Hemolysis |
| Hemoglobin | ↓ | Microangiopathic hemolysis |
Screening Laboratory Tests in Evaluation of Bleeding
| Test | What it Measures | Pathway Assessed |
|---|---|---|
| Platelet Count | Quantitative platelet assessment | Primary hemostasis |
| Platelet Function Analyzer (PFA-100) / Closure Time | Global platelet function (replaces bleeding time) | Primary hemostasis |
| Prothrombin Time (PT) / INR | Factors VII, X, V, prothrombin (II), fibrinogen | Extrinsic + Common pathways |
| Activated Partial Thromboplastin Time (aPTT) | Factors XII, XI, IX, VIII, X, V, prothrombin, fibrinogen | Intrinsic + Common pathways |
| Thrombin Time (TT) | Fibrinogen quantity and quality; heparin effect | Final common step (fibrinogen → fibrin) |
| Fibrinogen assay | Quantitative; also qualitative (dysfibrinogenemia) | |
| D-dimer | Fibrin cross-link degradation products | Fibrinolysis; elevated in DIC, PE, DVT |
| Peripheral Blood Smear | Platelet morphology, schistocytes, giant platelets | |
| Mixing Study | Patient plasma + normal plasma (1:1) | Corrects if factor deficiency; does NOT correct if inhibitor present |
| Specific Factor Assays | Quantitative individual factor levels | Confirm specific hemophilia type |
| vWF Antigen | Quantity of vWF protein | vWD diagnosis |
| Ristocetin Cofactor Assay | vWF functional activity | vWD; ↓ in Type 1,2,3; absent in Type 3 |
| RIPA (Ristocetin-Induced Platelet Agglutination) | vWF-platelet interaction | ↓ in most vWD; ↑ in Type 2B |
| ADAMTS13 activity | Metalloprotease for ultra-large vWF multimers | Severely ↓ in TTP |
| Platelet aggregation studies | Response to ADP, collagen, epinephrine, ristocetin | Distinguish Glanzmann vs Bernard-Soulier |
Differential Patterns at a Glance
| Disorder | Platelet Count | PT | aPTT | TT | Bleeding Time/PFA | Key Special Test |
|---|---|---|---|---|---|---|
| Thrombocytopenia | ↓ | N | N | N | ↑ | Bone marrow biopsy, antiplatelet Ab |
| Glanzmann Thrombasthenia | N | N | N | N | ↑↑ | No aggregation with ADP/collagen; GpIIb/IIIa absent |
| Bernard-Soulier | N (giant platelets) | N | N | N | ↑↑ | No ristocetin agglutination; GpIb absent |
| Hemophilia A | N | N | ↑ | N | N | ↓ Factor VIII; corrects on mixing study |
| Hemophilia B | N | N | ↑ | N | N | ↓ Factor IX; corrects on mixing study |
| von Willebrand Disease | N | N | ↑ or N | N | ↑ | ↓ vWF antigen, ↓ ristocetin cofactor |
| Factor VII deficiency | N | ↑ | N | N | N | ↓ Factor VII |
| Vitamin K deficiency | N | ↑↑ | ↑ | N | N | Corrects with Vit K/FFP |
| Liver disease | ↓ | ↑ | ↑ | ↑ | ↑ | ↓ Fibrinogen; hepatic panel |
| DIC | ↓↓ | ↑↑ | ↑↑ | ↑↑ | ↑ | ↑↑ D-dimer, ↓ fibrinogen, schistocytes |
| ITP | ↓↓ | N | N | N | ↑ | ↑ Megakaryocytes in marrow; antiplatelet Ab |
| TTP | ↓ | N | N | N | ↑ | ↓ ADAMTS13 activity; schistocytes |
(N = Normal)
Clinical Pearl: Type of Bleeding Localizes the Defect
| Feature | Platelet/Vascular Disorder | Coagulation Factor Disorder |
|---|---|---|
| Bleeding site | Superficial (skin, mucosa) | Deep (muscles, joints) |
| Skin manifestation | Petechiae, purpura, ecchymoses | Large hematomas |
| Mucous membrane | Common (epistaxis, gum, GI, GU) | Rare |
| Hemarthrosis | Rare | Classic (hemophilia) |
| Onset after injury | Immediate (< 1 hour) | Delayed (hours later) |
| Response to pressure | Often stops with pressure | Does not stop |
Robbins, Cotran & Kumar Pathologic Basis of Disease | Goldman-Cecil Medicine, International Ed. | Henry's Clinical Diagnosis and Management by Laboratory Methods | Tintinalli's Emergency Medicine | Berek & Novak's Gynecology (for ITP classification)
Quality assurance in hematology lab for 20 marks
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have rich, authoritative content. Let me compile the comprehensive 20-mark answer.
Quality Assurance in the Hematology Laboratory
Definition and Overview
Quality Assurance (QA) is a systematic process encompassing all activities - from specimen collection to result reporting - that are designed to ensure that laboratory test results are accurate, reliable, reproducible, and clinically useful. In the hematology laboratory, QA is not limited to controlling analyzers; it covers the entire testing cycle.
Quality Control (QC) is a subset of QA focused specifically on monitoring the analytical performance of measurement procedures to verify they meet defined specifications.
The Testing Cycle: Three Phases of Error
All laboratory errors are categorized into three phases. Studies consistently show pre-analytical errors are most common (~46-68%), followed by post-analytical errors, with analytical errors being the least frequent (~7-13%).
PRE-ANALYTICAL → ANALYTICAL → POST-ANALYTICAL
(before testing) (during testing) (after testing)
I. Pre-Analytical Phase
This phase covers all steps before the specimen reaches the analyzer.
1. Patient Preparation
- Correct patient identification (two identifiers: name + date of birth or ID number)
- Appropriate timing: fasting state, time of collection relative to medication
- Avoiding diurnal variation (e.g., eosinophil counts higher in morning)
- Position effects: WBC and platelet counts can vary slightly with posture
2. Specimen Collection
- Correct anticoagulant: EDTA (K₂EDTA or K₃EDTA) tubes (lavender/purple top) are standard for CBC, differential, and platelet count
- EDTA chelates calcium, preventing coagulation without affecting cell morphology
- Citrate tubes (blue top) used for coagulation studies - 9:1 ratio of blood to citrate is critical
- Correct fill volume: Under-filled tubes change the blood:anticoagulant ratio
- Under-filled EDTA: EDTA excess causes RBC shrinkage → falsely low MCV and hematocrit
- Under-filled citrate: excess citrate → falsely prolonged PT/PTT
- Order of draw: Prevents cross-contamination between additives
- Venepuncture technique: Avoid prolonged tourniquet (>1 minute → haemoconcentration), traumatic venepuncture (haemolysis, platelet activation), and IV line contamination
3. Specimen Labeling and Transport
- Labels must be applied immediately after collection at the bedside
- Barcoding enables positive patient and specimen identification through the entire pre-analytical, analytical, and post-analytical journey; 2D barcodes (QR-type) preferred over 1D barcodes for error correction capability
- Transport at room temperature; avoid excessive heat, cold, or agitation
- Coagulation specimens must be tested within 4 hours at room temperature; if delayed, plasma should be separated and frozen at -20°C
4. Common Pre-Analytical Errors in Hematology
| Error | Effect |
|---|---|
| EDTA-induced pseudothrombocytopenia | Platelet clumping → falsely low platelet count (verify with peripheral smear or citrate tube) |
| Haemolysis | Falsely elevated WBC, MCV; interferes with Hb optical measurement |
| Lipaemia (turbidity) | Falsely elevated Hb by spectrophotometric methods |
| Clotted specimen | Falsely low counts; reject specimen |
| Delayed processing (>4h) | WBC morphology distortion; platelet satellitosis; lysis of fragile cells |
| Icterus (severe) | Interferes with Hb measurement |
| Cold agglutinins | Falsely low RBC, falsely high MCV and MCHC at room temperature; warm specimen to 37°C |
II. Analytical Phase
A. Instrumentation and Calibration
Modern automated hematology analyzers (e.g., Sysmex, Beckman Coulter, Abbott) measure CBC parameters using:
- Electrical impedance (Coulter principle): Cell counting and sizing
- Light scattering (laser/optical): Differential counting, reticulocyte analysis, platelet indices
- Fluorescence flow cytometry: Nucleated RBC, immature cell fractions
Calibration establishes the correct relationship between instrument signal and measurand value. Calibrators are traceable to reference methods. Key principles:
- Calibrate when installing new instrument, after major maintenance, when QC indicates systematic drift
- Calibrators must be commutable with patient samples
B. Internal Quality Control (IQC)
Definition: IQC involves measuring control samples with known target values alongside patient samples at regular intervals to verify the measurement procedure is performing correctly before results are released.
QC Materials:
- Commercially prepared, stabilized blood-based controls (e.g., XE-Alpha N/L/H levels for Sysmex analyzers)
- Three levels typically used: low, normal, and high - to monitor performance across the analytical measuring interval
- Same lot should be used for extended periods (≥1 year) to establish reliable interpretive criteria
Statistical Foundation:
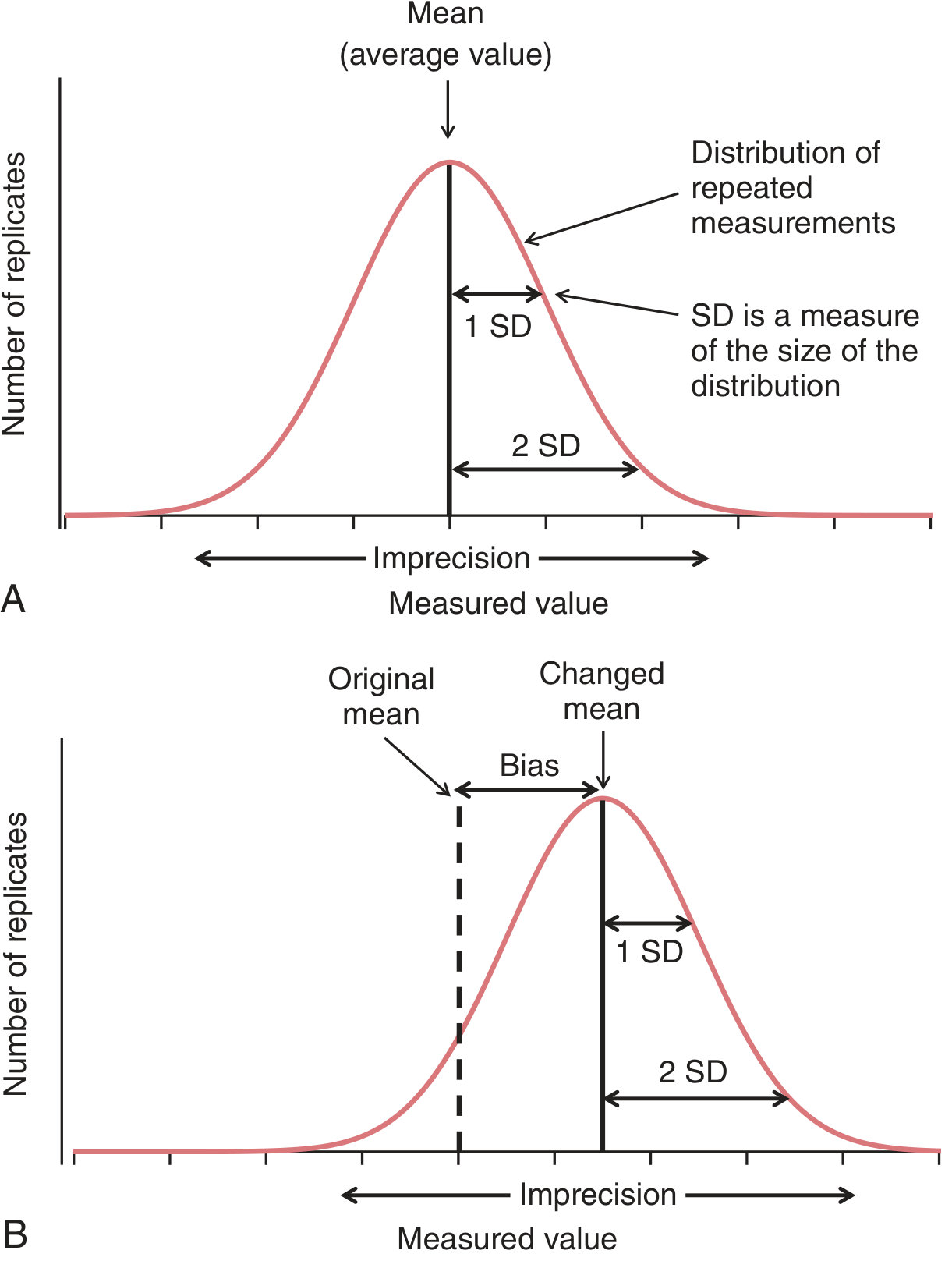
(A) Normal distribution of QC results around the mean; SD measures imprecision. (B) Calibration bias introduces a systematic shift - the new mean deviates from the established target value
Key statistical concepts:
- Mean (target value): Established by running the control ≥20 times
- Standard Deviation (SD): Reflects random imprecision; ±1 SD = 68.3%, ±2 SD = 95.4%, ±3 SD = 99.7% of all values
- Coefficient of Variation (CV%) = (SD/Mean) × 100: Allows comparison of imprecision across different analytes
- Bias: Systematic deviation from the true value; detected by calibration drift
C. Levey-Jennings Chart
The Levey-Jennings chart is the primary graphical tool for evaluating QC performance over time. It is an adaptation of the Shewhart industrial control chart.
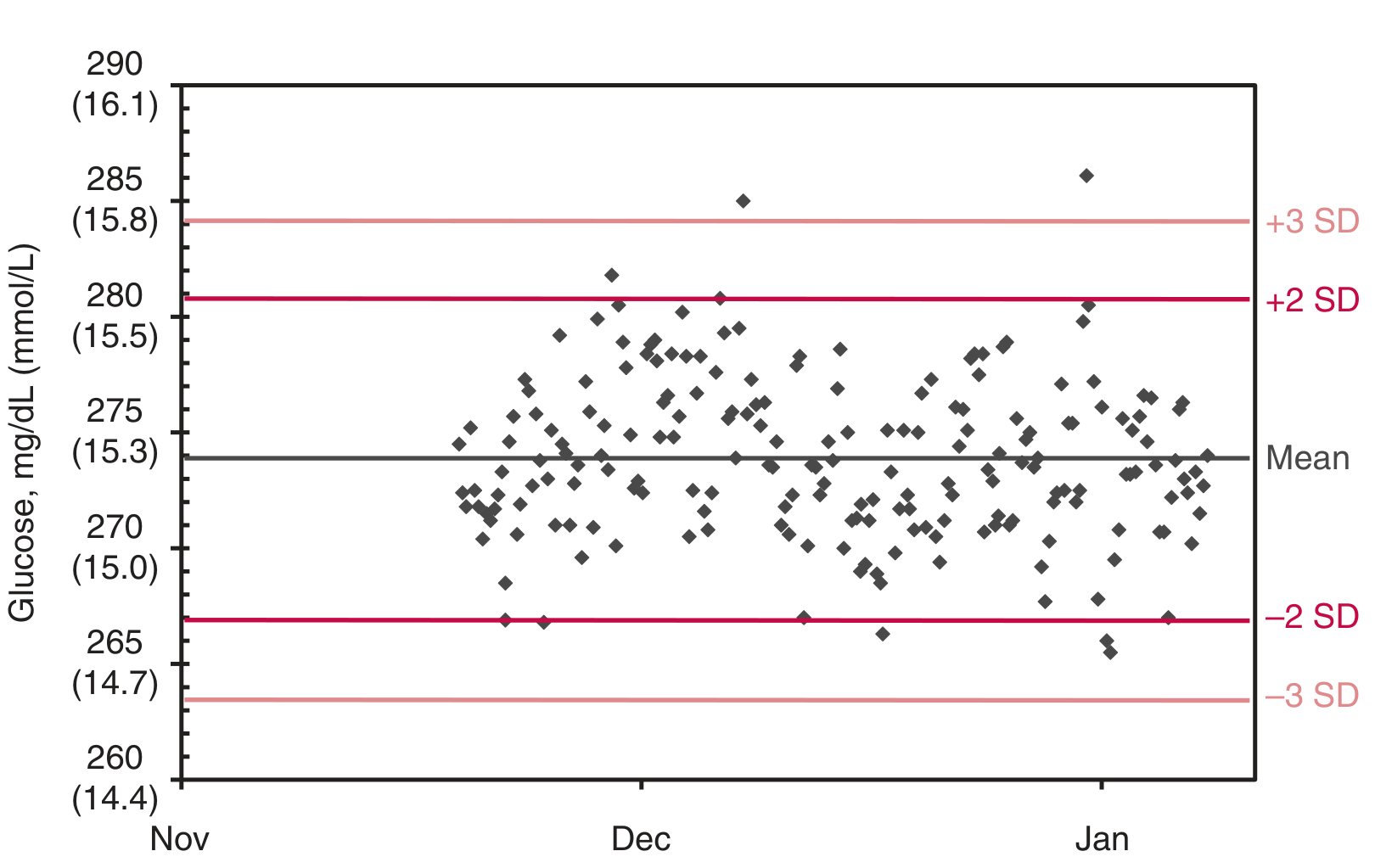
QC results plotted sequentially over time; horizontal lines at Mean, ±1 SD, ±2 SD, and ±3 SD. Results should fall randomly within ±2 SD for a stable process
Construction:
- X-axis: Time (sequential run number or date)
- Y-axis: QC result value
- Lines drawn at: Mean, ±1 SD, ±2 SD, ±3 SD
Interpretation:
- Results within ±2 SD: in control (acceptable)
- Results outside ±2 SD: warning (evaluate with rules)
- Results outside ±3 SD: rejection (immediate action)
D. Westgard Rules
Westgard rules are a set of statistical decision criteria applied to QC results to distinguish true analytical errors from random variation. They reduce false rejections while maximizing error detection.
| Rule | Symbol | Interpretation | Error Type Detected |
|---|---|---|---|
| 1₂s | Warning | 1 QC result > ±2 SD | Warning only - evaluate further |
| 1₃s | Rejection | 1 QC result > ±3 SD | Random error |
| 2₂s | Rejection | 2 consecutive results > ±2 SD (same side) | Systematic error (shift) |
| R₄s | Rejection | 1 result >+2 SD AND another >-2 SD in same run | Random error |
| 4₁s | Rejection | 4 consecutive results > ±1 SD (same side) | Systematic error (trend) |
| 10ₓ | Rejection | 10 consecutive results on same side of mean | Systematic error (drift) |
Practical application:
- 1₂s is the initial warning trigger
- If warning triggered, apply all other rules
- If any rejection rule is violated → do not release results → investigate and correct
- Trends (gradual drift toward one side) indicate reagent deterioration, calibration drift, or temperature changes
- Shifts (sudden jump to one side) indicate calibration change, reagent lot change, or instrument malfunction
E. Sigma Metrics and Six Sigma in the Laboratory
The Sigma metric quantifies how well a measurement procedure performs relative to clinical requirements:
Sigma = (TEa − Bias) / SD
Where:
- TEa = Total allowable error (based on clinical requirements, e.g., CLIA, biological variation)
- Bias = Systematic error (estimated from EQA)
- SD = Imprecision
| Sigma Value | Interpretation | QC Strategy Required |
|---|---|---|
| ≥6 | Excellent performance | Minimum QC (single control, 1₃s rule) |
| 4-5 | Good | Moderate QC (2 controls, 1₂s/1₃s) |
| 3-4 | Marginal | Tight QC (3+ controls, multirule) |
| <3 | Unacceptable | Improve measurement procedure |
Six Sigma = < 3.4 defects per million opportunities - the ideal target for clinical laboratory testing.
F. Moving Average / Bull's Algorithm (Patient-Based QC)
Unique to hematology: The XB method (Moving Average of Red Cell Indices) - also called Bull's algorithm - uses the running average of patient MCV, MCH, and MCHC from the last 20 samples to continuously monitor analyzer performance.
- If the moving average drifts outside ±3% of target, it signals analyzer malfunction
- Advantage: detects systematic errors in real time between scheduled QC runs
- Limitation: assumes patient population is stable; unreliable during population shifts (e.g., post-transfusion, ICU patients)
III. Post-Analytical Phase
1. Reference Intervals
- Validated reference intervals specific to the laboratory's population (age, sex, ethnicity)
- Pediatric values differ markedly from adult values (e.g., neonatal Hb 14-20 g/dL)
- Must be reviewed when changing analyzers or reagent lots
2. Critical Values (Panic Values)
- Predefined thresholds requiring immediate clinician notification:
| Parameter | Critical Low | Critical High |
|---|---|---|
| Haemoglobin | <7 g/dL | >20 g/dL |
| WBC | <2 × 10⁹/L | >30 × 10⁹/L |
| Platelets | <50 × 10⁹/L | >1000 × 10⁹/L |
| INR | - | >5.0 |
- All critical value notifications must be documented (time, person notified, person reporting)
3. Delta Checks
- Comparison of current result with previous result for the same patient
- Flags results that differ by more than a defined threshold (delta limit)
- Detects: specimen identification errors (wrong patient), analytical errors, genuine acute clinical changes
- Example: Hb delta check >2 g/dL → verify specimen identity before releasing result
4. Result Review and Smear Preparation
- Automated analyzers flag abnormal results (morphology flags, immature cell flags) requiring manual peripheral blood smear review
- ICSH (International Council for Standardization in Haematology) criteria define when smear review is mandatory
- Examples of flags requiring smear: blasts, atypical lymphocytes, left shift, NRBC, platelet clumps
5. Turnaround Time (TAT)
- Defined targets for routine and urgent/STAT testing
- TAT monitoring is a key performance indicator (KPI)
- Common benchmark: STAT CBC reported within 60 minutes of receipt
6. Result Reporting and Transcription
- Laboratory Information System (LIS) integration minimizes transcription errors
- Verified, authorized results only
- Corrected reports must be issued with documentation when errors are found after release
IV. External Quality Assessment (EQA) / Proficiency Testing (PT)
Definition: EQA is a process in which control samples from an independent external organization are distributed to participating laboratories; expected values are unknown at the time of testing. Results are compared with peer group or reference method targets.
Purpose:
- Assesses trueness (systematic bias of a method)
- Allows inter-laboratory and inter-method comparison
- Identifies outliers and systematic problems not detectable by IQC
- Required for laboratory accreditation (ISO 15189, CAP, NABL)
Process:
- External agency (e.g., UK NEQAS, CAP, RCPAQAP) sends lyophilized or stabilized samples
- Laboratory tests them like routine samples
- Results returned to agency; laboratory receives performance report
- Unacceptable results trigger root cause analysis and corrective action
IQC vs EQA:
| Feature | Internal QC (IQC) | External QA (EQA/PT) |
|---|---|---|
| Frequency | Every run/daily | Monthly/quarterly |
| Target values | Known in advance | Unknown until results submitted |
| Purpose | Detect analytical errors in real time | Detect systematic bias; inter-lab comparison |
| Source | In-house/commercial | Independent external agency |
| Action | Do not release results if fail | Root cause analysis; corrective action |
V. Personnel Competency Assessment
Per regulatory requirements (CLIA, CAP):
- Six elements of competency must be documented for each employee for each test:
- Direct observation of routine patient test performance
- Monitoring of recording and reporting of results
- Review of intermediate test results, QC records, and PT results
- Direct observation of preventive maintenance and function checks
- Assessment of problem-solving skills
- Testing of unknown specimens (where applicable)
- Competency must be documented within the first 6 months of testing, then annually, and whenever methodology or instruments change
VI. Hematology-Specific QA Considerations
| Aspect | Hematology-Specific Issue | QA Measure |
|---|---|---|
| EDTA pseudothrombocytopenia | Platelet clumping in EDTA | Peripheral smear review; repeat in citrate tube |
| Cold agglutinins | Falsely low RBC, high MCV/MCHC | Warm specimen to 37°C; compare pre/post warming |
| Morphology review criteria | Automated flags for blasts, atypical cells | ICSH-defined smear criteria; trained morphologist review |
| Bull's algorithm (XB method) | Real-time patient-based RBC index monitoring | Flag if MCV/MCH/MCHC moving average drifts >3% |
| Reticulocyte counting | High CV in manual counting (~25%) vs automated | Use automated reticulocyte count with fluorescence; QC at two levels |
| Coagulation testing | Pre-analytical sample quality critical | 9:1 blood:citrate ratio; test within 4 hours; reject clotted/haemolysed samples |
| Peripheral blood smear staining | Stain quality affects morphology interpretation | Positive staining control included daily; check Romanowsky stain pH |
VII. Laboratory Accreditation Standards
Key standards governing QA in hematology labs:
- ISO 15189:2022 - International standard for medical laboratory quality and competence
- CLSI (Clinical and Laboratory Standards Institute) - H20 series for hematology QC
- CAP (College of American Pathologists) - Accreditation checklists
- NABL (National Accreditation Board for Testing and Calibration Laboratories) - India
- CLIA (Clinical Laboratory Improvement Amendments) - USA federal regulation
Summary: Key Components of QA in Hematology
QA IN HEMATOLOGY LABORATORY
│
├── PRE-ANALYTICAL
│ ├── Patient identification (barcoding, 2 identifiers)
│ ├── Correct specimen collection (EDTA, fill volume, order of draw)
│ ├── Transport and storage conditions
│ └── Rejection criteria (clotted, haemolysed, wrong tube)
│
├── ANALYTICAL
│ ├── Calibration (traceable to reference methods)
│ ├── IQC (3-level controls, Levey-Jennings chart)
│ ├── Westgard Rules (1₃s, 2₂s, R₄s, 4₁s, 10ₓ)
│ ├── Bull's Algorithm (XB moving average for RBC indices)
│ └── Sigma metrics (TEa - bias / SD)
│
├── POST-ANALYTICAL
│ ├── Reference intervals (validated, population-specific)
│ ├── Critical values (immediate notification + documentation)
│ ├── Delta checks (intra-patient comparison)
│ ├── Peripheral smear review (flagged samples)
│ └── TAT monitoring
│
└── EXTERNAL
├── EQA/Proficiency Testing (independent agency)
├── Personnel competency documentation (6 elements, annual)
└── Accreditation (ISO 15189, CAP, NABL, CLIA)
Tietz Textbook of Laboratory Medicine, 7th Ed. (Chapters 2 & 6) | Henry's Clinical Diagnosis and Management by Laboratory Methods | Quick Compendium of Clinical Pathology, 5th Ed.
Transfusion transmitted infections in blood banking, 20 marker
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have rich, comprehensive content from multiple authoritative sources. Let me now compile the full 20-mark answer.
Transfusion Transmitted Infections (TTIs) in Blood Banking
Introduction
Transfusion Transmitted Infections (TTIs) are infections that are acquired by a recipient as a direct consequence of blood transfusion or administration of blood components/products. Despite modern screening measures, the blood supply cannot be rendered 100% safe because of the window period - the interval between infection of the donor and the point at which that infection is detectable by current laboratory assays.
Blood safety is maintained through a multi-layered strategy:
- Donor selection and deferral (questionnaire-based)
- Laboratory testing of donated units
- Pathogen reduction/inactivation technologies
- Hemovigilance and post-donation surveillance
The Window Period - Central Concept
Definition: The window period is the time interval between the moment a donor becomes infected and when the infection can be reliably detected by a given screening test. During this period, blood from an infected donor may test negative and be inadvertently released, posing a transmission risk.
Key principle: NAT (Nucleic Acid Testing) substantially reduces the window period compared to serological assays (antibody/antigen tests), as viral nucleic acids appear in the bloodstream earlier than detectable antibody/antigen responses.
| Test Type | Detects | Window Period Advantage |
|---|---|---|
| Serology (EIA/ChLIA) | Antibodies or antigens | Longer window (weeks to months) |
| NAT (PCR/TMA) | Viral RNA/DNA | Shorter window (days) |
Mandatory Testing (Per National Blood Safety Policy - India/WHO)
As per the national blood safety policy, testing of every unit of blood is mandatory for:
- HIV (Types 1 and 2)
- Hepatitis B virus (HBV)
- Hepatitis C virus (HCV)
- Syphilis (Treponema pallidum)
- Malaria (Plasmodium species)
Park's Textbook of Preventive and Social Medicine
Additionally, in developed countries (USA, UK, Europe), testing also includes:
6. HTLV-I/II
7. West Nile Virus (WNV) - NAT in endemic seasons
8. Chagas disease (Trypanosoma cruzi) - endemic regions
9. Babesia species - endemic states (USA)
Residual Risk and Window Periods (Goldman-Cecil Medicine, Table 162-3)
| Infection | Test Used | Window Period (Days) | Residual Risk |
|---|---|---|---|
| HIV | MP-NAT | 9 | ~1 : 1,800,000 |
| HIV | EIA (antibody) | 21 | - |
| HCV | MP-NAT | 7 | ~1 : 1,600,000 |
| HCV | EIA (antibody) | 51-58 | - |
| HBV | HBsAg | 30-38 | ~1 : 300,000 |
| HBV | NAT (individual) | 15-34 | ~1 : 1,500,000 |
| HTLV-I/II | Antibody EIA | ~80 | ~1 : 3,300,000 |
| Syphilis | Antibody | - | 1 reported case in USA in 50 years |
| Chagas | Antibody | - | 7 reported cases in USA |
| Bacterial (all) | Culture | - | 1 : 3,000 |
| Bacterial (platelets) | Culture | - | 1 : 20,000 |
(MP-NAT = mini-pool nucleic acid testing)
I. Viral Transfusion Transmitted Infections
1. Human Immunodeficiency Virus (HIV)
History: HIV emerged in the early 1980s when blood banking was unprepared. At the peak, 1 in 100 units in some US cities contained HIV. With current screening practices, risk has fallen to ~1 in 2 million units.
Screening tests:
- Antibody detection: Chemiluminescent immunoassay (ChLIA) for anti-HIV-1 (groups M, N, O) and anti-HIV-2; detects antibodies ~3 weeks post-infection
- NAT (HIV-1 RNA): Reduces window period to ~9-10 days
- Combined antigen/antibody (4th generation) assays detect both p24 antigen and antibodies
Donor deferral:
- High-risk behaviors: MSM within 3 months, sex workers, IV drug users
- History of positive HIV test → permanent deferral
- Recipients of antiretroviral PrEP/PEP → deferred for 3 months after last dose
Clinical consequence in recipient: AIDS; fatal if untreated; profound immunodeficiency
2. Hepatitis B Virus (HBV)
Epidemiology: >300 million carriers worldwide; highly prevalent in developing countries. Acute infection often asymptomatic; may progress to chronic hepatitis, cirrhosis, hepatocellular carcinoma.
Screening tests:
- HBsAg (Hepatitis B surface antigen): Window period 35-40 days; first introduced 1972
- Anti-HBc (core antibody): Can be positive as early as 1 week post-infection; high false-positive rate (~1% of donors); used to detect occult HBV infection (OBI)
- HBV NAT (DNA): Reduces window to 15-34 days (individual testing) or 40-50 days (mini-pool)
- Confirmatory: HBsAg neutralization
Donor deferral: Contact/household/sexual exposure to hepatitis → deferred 12 months
Special note: Occult HBV Infection (OBI) - donors negative for HBsAg but positive for HBV DNA - can still transmit HBV; hence the importance of anti-HBc and NAT testing.
Prevention bonus: Global HBV vaccination programs have successfully reduced HBV prevalence in donor populations.
3. Hepatitis C Virus (HCV)
Epidemiology: Progresses to chronic carrier state in 80-85% of infected individuals; major cause of cirrhosis, hepatocellular carcinoma, and liver failure.
Screening tests:
- Anti-HCV antibody (EIA/ChLIA): Window period 51-70 days post-infection
- HCV NAT (RNA): Shortens window to 7-8 days - most significant advance in HCV blood safety
- NAT introduced for HCV in blood banking in 2000
Residual risk: ~1 in 1.6-2.6 million donations (post-NAT era)
Note: HCV prevalence in donor populations has increased in recent years due to the opioid crisis, making continued vigilance essential.
4. Human T-Lymphotropic Virus (HTLV-I/II)
Disease associations:
- HTLV-I: Adult T-cell leukemia/lymphoma (ATL); HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP)
- HTLV-II: Generally less pathogenic; no significant disease in immunocompetent individuals
- Both: lymphadenopathy
Screening: Combination ChLIA for anti-HTLV-I/II antibodies; no NAT available
- Window period: ~80 days
- Residual risk: ~1 in 2-3.3 million units
- Donors testing reactive → no re-entry protocol (permanent deferral)
Key note: HTLV is cell-associated (transmitted via infected lymphocytes), so leukoreduction of blood products significantly reduces - though does not eliminate - transmission risk.
5. Cytomegalovirus (CMV)
Epidemiology: Seroprevalence 50-85% in general adult population; previously considered the most common TTI.
Transmission: Via CMV-positive white blood cells (cell-associated virus)
Clinical significance:
- Immunocompetent recipients: Usually asymptomatic or mild self-limited illness
- High-risk recipients (neonates, HIV patients, organ/stem cell transplant recipients): Severe multiorgan failure - hepatitis, pneumonitis, retinitis, GI disease, marrow failure
Prevention:
- Use of CMV-seronegative blood products for at-risk patients
- Leukoreduction (LR): Significantly reduces transmission risk (from 1-3% to 0.023%); reduces but does not eliminate risk completely
Residual risk with leukoreduction: ~0.023%
6. West Nile Virus (WNV)
- Mosquito-borne flavivirus; emerged in USA 1999; peak epidemic 2002-2003 (23 transfusion-transmitted cases)
- Clinical: Mild febrile illness in most; encephalitis/meningitis in immunosuppressed
- NAT testing introduced in 2003 dramatically reduced cases to only 13 since 2003
- Testing strategy: Mini-pool NAT; switch to individual donor NAT during seasonal outbreaks
7. Other Viruses
| Virus | Notes |
|---|---|
| Parvovirus B19 | Can cause aplastic crises in immunosuppressed, sickle cell patients; not routinely screened |
| Hepatitis E Virus (HEV) | Increasingly recognized as TTI; can cause chronic hepatitis in immunocompromised; some countries now screen |
| Zika Virus | Mosquito-borne flavivirus; NAT screening implemented in high-risk areas; causes microcephaly in neonates |
| SARS-CoV-2 | No confirmed transfusion transmission to date; donor deferral if recently exposed |
| Hepatitis A | Extremely rare via transfusion; short viremia, usually symptomatic at donation |
| EBV (Epstein-Barr) | Cell-associated; leukoreduction reduces risk; rarely clinically significant via transfusion |
II. Bacterial Contamination
Bacterial contamination is the most common infectious complication of transfusion and carries greater mortality risk than all viral TTIs combined.
Sources of contamination:
- Donor bacteremia at time of donation (transient, e.g., from dental procedures, skin infections)
- Skin flora introduced at venepuncture - most common; skin organisms (Staphylococcus, Bacillus species) are drawn into collection tubing with the initial blood flow
Risk by blood component:
| Component | Storage Conditions | Risk | Common Organisms |
|---|---|---|---|
| Platelets | 20-24°C (room temp) with agitation; 5-7 days | Highest: ~1:20,000 | Staphylococcus aureus, Streptococcus, Gram-negative rods |
| Red blood cells | 4°C (refrigeration); up to 42 days | Lower: ~1:250,000-1:10 million | Cold-tolerant Gram-negatives: Yersinia enterocolitica, Serratia, Pseudomonas |
| Fresh Frozen Plasma | Frozen (-18°C) | Negligible | - |
Why platelets are higher risk: Room temperature storage promotes exponential bacterial replication; prolonged shelf life (5-7 days) allows inoculum to grow to clinical levels.
Why cold storage RBCs pose risk from cold-tolerant bacteria: Yersinia enterocolitica and Pseudomonas species proliferate at 4°C → septic shock upon transfusion.
Prevention of bacterial contamination:
- Diversion pouch: First 20-40 mL of blood diverted into a separate chamber to exclude skin-plug contamination
- Skin antisepsis: Rigorous cleaning with chlorhexidine-isopropanol
- Sterile collection technique
- Culture-based testing of platelets (mandatory in USA since 2004):
- BACTEC blood culture system: 8 mL sampled ≥24h after collection
- Growth detected by pH change, CO₂ production
- Point-of-issue rapid testing (prior to platelet transfusion):
- Colorimetric peptidoglycan detection
- Lateral flow immunoassay for lipoteichoic acid/LPS
- Pathogen inactivation (see below)
III. Parasitic Transfusion Transmitted Infections
1. Malaria (Plasmodium species)
- Most important parasitic TTI worldwide (especially in endemic regions - Sub-Saharan Africa, South Asia)
- Mandatory screening in India - microscopical examination of all donor blood + malarial antibody ELISA
- Donor deferral: Travel to malaria-endemic areas (12 months deferral); history of malaria (3 years deferral)
- RBCs stored at 4°C: Plasmodium trophozoites can survive for the shelf life of blood
- Clinical consequence: Transfusion-transmitted malaria can be fatal, especially in non-immune recipients
2. Chagas Disease (Trypanosoma cruzi)
- Endemic in Latin America; significant concern with immigration patterns
- Testing with Chagas antibody EIA now performed in USA and many European countries
- 7 reported cases in USA prior to testing; none since testing implemented
- Trypanosomes survive in refrigerated RBCs for weeks
3. Babesiosis (Babesia microti)
- Tick-borne protozoan; endemic in northeastern USA
- Causes hemolytic anemia; life-threatening in asplenic/immunocompromised patients
- NAT testing now required in endemic US states
- Donor deferral: History of Babesia → deferred; re-entry possible after 2 years if testing negative
4. Toxoplasma gondii
- Rare TTI; mainly in immunocompromised recipients
- No routine screening; managed through donor history questionnaire
IV. Prion Diseases
Variant Creutzfeldt-Jakob Disease (vCJD)
- Caused by abnormal prion proteins (PrPSc); related to bovine spongiform encephalopathy (BSE/"mad cow disease")
- UK epidemic in 1980s-1990s; 4 confirmed transfusion-transmitted cases in the UK
- No available screening test for prions in blood
- Managed entirely through donor deferral:
- Residence in UK >3 cumulative months (1980-1996) → permanent deferral
- Residence in France/Ireland >5 years (1980-present) → permanent deferral
- Fatal neurodegenerative disease; incubation period years to decades
- Prions cannot be inactivated by standard pathogen reduction technologies
V. Pathogen Reduction / Inactivation Technologies (PRT)
Given limitations of testing (window period, emerging pathogens), pathogen inactivation offers a complementary layer of safety:
| Technology | Target | Blood Component |
|---|---|---|
| Solvent-Detergent (SD) | Lipid-enveloped viruses only | Plasma products; pooled plasma |
| Psoralen + UV-A light (INTERCEPT) | Nucleic acid damage → inactivates bacteria, enveloped + non-enveloped viruses, parasites; NOT prions | Platelets, plasma |
| Riboflavin + UV light (Mirasol) | Nucleic acid damage | Platelets, plasma, RBCs (developing) |
| UV-C light | Nucleic acid damage | Platelets |
Key principle: Photochemical pathogen inactivation technologies target nucleic acids; since prions lack nucleic acids, they cannot be inactivated by these methods.
VI. Donor Selection: The First Line of Defense
The Donor History Questionnaire (DHQ) is the cornerstone of blood safety. No laboratory test can substitute entirely for donor self-exclusion.
Key Deferral Categories:
| Deferral Type | Examples |
|---|---|
| Permanent | History of HIV, HTLV, vCJD risk, Chagas, hemophilia, CJD |
| 12-month deferral | Incarceration >72h, tattoo/body piercing, blood transfusion/transplant, sexual contact with high-risk individuals, syphilis/gonorrhea treatment |
| 3-month deferral | MSM activity, sex worker contact, HIV PrEP/PEP |
| 6-month deferral (India) | Recent tattoo, ear/nose piercing |
| Travel-based | UK residence 1980-1996 (vCJD), malaria-endemic areas (12 months) |
| 24-hour deferral | Recent fever, dental procedure, minor surgery |
Key principle: Professional/paid blood donation is associated with higher TTI rates. Voluntary, non-remunerated blood donation is the WHO-recommended standard. In India, professional blood donation was banned from 1 January 1998.
VII. Summary: Multi-Layered Blood Safety Strategy
BLOOD SAFETY PYRAMID
│
├── 1st Layer: Voluntary non-remunerated donors (lowest risk population)
│
├── 2nd Layer: Donor History Questionnaire (DHQ) - behavioral/travel deferral
│
├── 3rd Layer: Mandatory Serological Screening (ELISA/ChLIA)
│ HIV 1/2, HBsAg, anti-HCV, anti-HBc, anti-HTLV, RPR/TPHA,
│ Malaria antibody, Chagas Ab, WNV NAT (seasonal)
│
├── 4th Layer: NAT (Nucleic Acid Testing - PCR/TMA)
│ HIV RNA, HCV RNA, HBV DNA, WNV RNA
│ → Dramatically shortens window period
│
├── 5th Layer: Pathogen Inactivation (for platelets, plasma)
│ INTERCEPT, Mirasol systems
│
├── 6th Layer: Leukoreduction (for CMV, HTLV risk reduction)
│
└── 7th Layer: Hemovigilance (post-transfusion surveillance, lookback)
Key Table: Comparison of TTIs
| Agent | Type | Mandatory Screening | Window (NAT) | Residual Risk | Clinical Consequence |
|---|---|---|---|---|---|
| HIV 1/2 | Retrovirus | Yes | 9-10 days | 1:1.8 million | AIDS |
| HBV | Hepadnavirus | Yes | 15-34 days | 1:300K-1.5M | Hepatitis → cirrhosis → HCC |
| HCV | Flavivirus | Yes | 7-8 days | 1:1.6-2.6 million | Chronic hepatitis → cirrhosis |
| HTLV I/II | Retrovirus | Yes (developed countries) | ~80 days | 1:3.3 million | ATL, HAM/TSP |
| CMV | Herpesvirus | Serostatus only | N/A | 1-3% (all donors) | Severe in immunocompromised |
| Syphilis | Treponema pallidum | Yes | N/A | Extremely rare | Rarely transmitted (spirochete fragile) |
| Malaria | Plasmodium spp. | Yes (India/developing) | N/A | Significant in endemic areas | Hemolysis, cerebral malaria |
| Bacteria | Various | Platelets only | N/A | 1:20,000 (platelets) | Septic shock |
| Babesia | Protozoan | NAT (endemic areas) | N/A | Rare (screened) | Hemolytic anemia |
| Chagas | Trypanosoma cruzi | Yes (USA, Europe) | N/A | Rare (screened) | Cardiomyopathy |
| vCJD | Prion | None | N/A | 4 cases worldwide | Fatal neurodegeneration |
Tietz Textbook of Laboratory Medicine, 7th Ed. | Barash, Cullen & Stoelting's Clinical Anesthesia, 9th Ed. | Goldman-Cecil Medicine, International Ed. | Park's Textbook of Preventive and Social Medicine
Paroxysmal nocturna hemoglobinuria, 10 marker
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the complete 10-mark answer.
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Definition
Paroxysmal Nocturnal Hemoglobinuria (PNH) is a rare, acquired, clonal hematopoietic stem cell disorder characterized by the production of abnormal erythrocytes, granulocytes, and platelets that are exquisitely susceptible to complement-mediated lysis. It is the only hemolytic anemia caused by an acquired (somatic) genetic defect - as opposed to inherited red cell disorders.
- Incidence: 2-5 per million (annual)
- Age: Any age; most common between 10-50 years; mean age at diagnosis ~34 years
- Sex: Equal male:female ratio; no family history (acquired)
Pathogenesis / Molecular Basis
Step 1: The PIGA Mutation
A somatic mutation in the PIGA (Phosphatidylinositol Glycan class A) gene occurs in a single pluripotent hematopoietic stem cell:
- PIGA is located on the short arm of the X chromosome (Xp22.1)
- PIGA encodes an enzyme essential for the early step in GPI (Glycosylphosphatidylinositol) biosynthesis
- Because PIGA is X-linked, a single mutation in the active X allele is sufficient to abolish GPI synthesis in that cell and all its clonal descendants
- The mutation affects cells of all hematopoietic lineages (red cells, white cells, platelets)
- ~150 different PIGA mutations have been identified, most causing total loss of GPI production
Step 2: GPI Anchor Deficiency
GPI is a glycolipid molecule that anchors numerous proteins to the outer surface of the plasma membrane via a peptide bond to their C-terminal amino acid. Without functional GPI, these proteins are absent from the cell surface.
Step 3: Loss of Complement Regulatory Proteins
Three critical GPI-anchored complement regulatory proteins are absent from PNH cells:
| Protein | Also Known As | Function |
|---|---|---|
| CD59 | Protectin / MIRL (Membrane Inhibitor of Reactive Lysis) | Most important - inhibits polymerization of C9, preventing assembly of the Membrane Attack Complex (MAC = C5b-9) |
| CD55 | DAF (Decay Accelerating Factor) | Inhibits C3/C5 convertases of both classical and alternative complement pathways |
| C8-binding protein | Homologous Restriction Factor | Restricts MAC formation |
Other GPI-linked proteins also deficient: CD14, CD16a (FcγRIII), CD24, CD58, acetylcholinesterase (AChE), leukocyte alkaline phosphatase (LAP)
Step 4: Complement-Mediated Lysis
Without CD55 and CD59, the alternative complement pathway activates spontaneously on the red cell surface (spontaneous C3 tick-over), assembles the MAC (C5b-9), and produces intravascular hemolysis.
Triggers that accelerate lysis:
- Sleep (mild respiratory acidosis → slight drop in pH → increased complement activation)
- Infection (activates complement via classical pathway)
- Surgery, physical stress, blood transfusion
- Acidosis, hypoxia
PNH Erythrocyte Types (Henry's)
| Type | GPI Level | Complement Sensitivity |
|---|---|---|
| Type I | Normal | Normal sensitivity |
| Type II | Partial deficiency | 3-5× increased sensitivity |
| Type III | Complete absence | 15-25× increased sensitivity |
Coexistence of Type II and III indicates two separate mutant clones.
Why Does the PNH Clone Expand?
Normal individuals harbor small numbers of PIGA-mutant hematopoietic cells (~1 in 50,000 RBCs) but these do not expand. In PNH, a second step - likely an autoimmune process - allows the clone to expand:
- PNH is frequently associated with aplastic anemia (AA), which has an autoimmune basis
- T cells destroying GPI-positive stem cells spare GPI-negative (PNH) stem cells → selective clonal expansion
- This explains the clinical overlap and transitions between PNH and AA
Clinical Features
1. Hemoglobinuria (Classic but not the most common presentation)
- Dark, port wine-colored urine (classically in the morning after overnight sleep)
- Only occurs in ~25% of patients during paroxysms
- Results from the mild acidosis during sleep activating complement
- Hemosiderinuria (urinary iron loss as hemosiderin) is almost constantly present - more reliable sign
2. Chronic Intravascular Hemolytic Anemia
- The dominant feature; usually mild to moderate
- Normocytic or hypochromic microcytic (due to chronic iron loss in urine)
- Reticulocytosis less than expected for degree of anemia
- Direct Antiglobulin Test (Coombs) is NEGATIVE - cardinal feature distinguishing PNH from autoimmune hemolytic anemia
3. Thrombosis - Leading Cause of Death
- Occurs in ~40% of patients; venous in 85% of cases
- Unusual sites:
- Budd-Chiari syndrome (hepatic vein thrombosis) → hepatomegaly, ascites
- Portal vein thrombosis
- Cerebral vein thrombosis
- Mesenteric vein thrombosis
- Mechanism: CD59-deficient platelets → phosphatidylserine externalization → prothrombinase complex formation; free hemoglobin scavenges nitric oxide (NO) → platelet aggregation and endothelial dysfunction
4. Smooth Muscle Dystonia (due to NO scavenging by free hemoglobin)
- Dysphagia (esophageal spasm)
- Abdominal pain/colic (~1/3 of patients)
- Erectile dysfunction in males
5. Bone Marrow Failure
- Pancytopenia: Neutropenia (in 3/5 patients) and thrombocytopenia (in 2/3 patients) are common
- Bone marrow may be hypocellular or show erythroid hyperplasia
- ~1/3 of cases evolve into aplastic anemia
6. Iron Deficiency
- Chronic urinary iron loss (hemosiderin in urine → hemosiderinuria)
- Coombs-negative hemolytic anemia + iron deficiency = suspect PNH
7. Transformation to Myeloid Neoplasm
- ~5% of patients eventually develop AML or myelodysplastic neoplasm
Diagnosis
1. Gold Standard: Flow Cytometry (Current Standard)
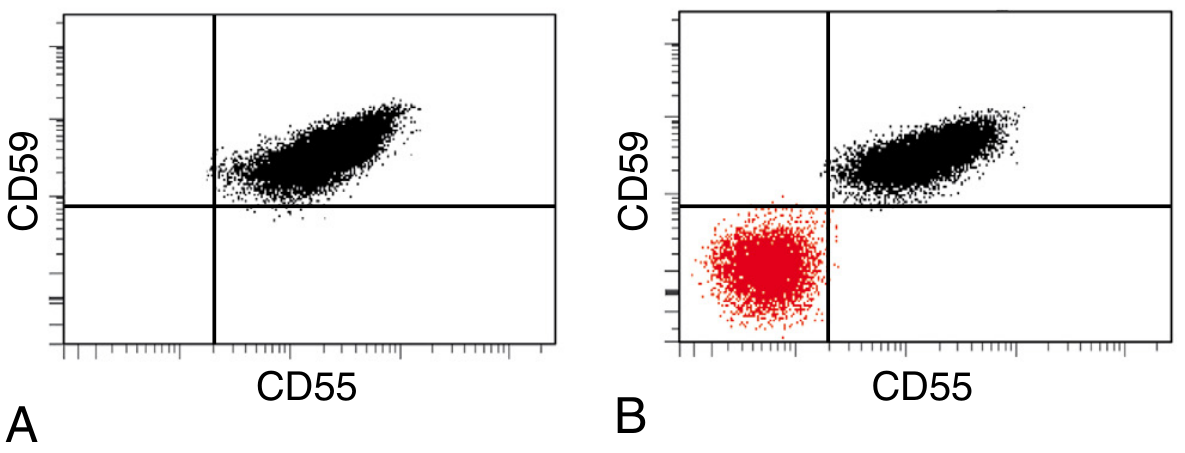
PNH diagnosis by flow cytometry: (A) Normal - all red cells in upper-right quadrant expressing both CD55 and CD59. (B) PNH - distinct population (red) in lower-left quadrant, negative for both CD55 and CD59
Flow cytometry panels:
- Red cells: CD59 (most sensitive for RBCs)
- Granulocytes: CD24, CD16, CD66b (best for small clones - not affected by hemolysis)
- Monocytes: CD14
- FLAER (Fluorescent Aerolysin Variant): Bacterial protein that binds directly to GPI anchor; most reliable and sensitive reagent for studying GPI-linked antigens on leukocytes; preferred for diagnosing PNH on granulocytes and monocytes
Clone size is directly correlated with degree of intravascular hemolysis and thrombosis risk.
2. Historical Tests (Now Superseded)
| Test | Principle | Why Replaced |
|---|---|---|
| Ham's Test (Acidified Serum Test) | Patient RBCs lysed by acidified normal serum (pH 6.2) - PNH cells lyse, normal cells don't | Low sensitivity; requires careful technique |
| Sugar Water Test (Sucrose Hemolysis) | Low ionic strength sucrose solution activates complement → PNH cells lyse | Screening test; less specific |
Both replaced by flow cytometry which is far more sensitive, specific, and quantitative.
3. Routine Laboratory Findings
| Test | Finding in PNH |
|---|---|
| Hemoglobin | Decreased (normocytic or microcytic/hypochromic) |
| Reticulocyte count | Elevated but less than expected |
| WBC | Decreased (neutropenia) |
| Platelets | Decreased (thrombocytopenia) |
| Direct Coombs test | NEGATIVE (key negative finding) |
| LDH | Elevated (marker of intravascular hemolysis) |
| Haptoglobin | Decreased/absent (free Hb binds haptoglobin) |
| Serum bilirubin (indirect) | Elevated |
| Plasma hemoglobin | Elevated (pink/red plasma) |
| Urinalysis | Hemoglobinuria (dark brown urine) + Hemosiderinuria (Prussian blue stain positive) |
| Serum iron, ferritin | Decreased (iron deficiency from chronic urinary loss) |
| Bone marrow | Hypercellular (erythroid hyperplasia) or hypocellular (if AA overlap) |
PNH Classification (Clinical Categories)
| Category | Features |
|---|---|
| Classic PNH | Dominant intravascular hemolysis; large clone; no marrow failure |
| PNH in setting of another bone marrow disorder | PNH clone + aplastic anemia or MDS; cytopenias predominant |
| Subclinical PNH | Small clone (<1%); no hemolysis; found incidentally in AA (50-60%) and MDS (15-20%) |
Treatment
1. Complement Inhibitors (Disease-Modifying Therapy)
Eculizumab (Anti-C5 monoclonal antibody) - First targeted therapy:
- Binds complement component C5, preventing cleavage to C5a+C5b → blocks MAC formation
- Eliminates intravascular hemolysis; reduces thrombosis risk by up to 90%
- Reduces/eliminates transfusion requirements
- Dose: 600 mg IV weekly × 4 weeks → 900 mg at week 5 → 900 mg every 2 weeks
- Side effect: Increased risk of Neisseria meningitidis infection (MAC normally defends against encapsulated organisms)
- Pre-treatment requirement: Vaccination against Meningococcus A, B, C (and Y/W) + prophylactic oral penicillin
Ravulizumab: Long-acting anti-C5; same mechanism; administered every 8 weeks (vs. 2 weeks for eculizumab)
Pegcetacoplan (Anti-C3 inhibitor): Newer; targets upstream C3, blocking both intravascular AND extravascular hemolysis; useful in patients with persistent anemia on C5 inhibitors
Note on eculizumab limitation: While blocking MAC formation (intravascular hemolysis), C3 fragments still opsonize PNH RBCs → Direct Coombs becomes positive → iatrogenic extravascular hemolysis occurs (spleen-mediated)
2. Supportive Treatment
- Iron and folate supplementation (to counter urinary losses and increased erythropoiesis)
- Blood transfusions (leukoreduced; for severe anemia)
- Anticoagulation for thrombotic episodes (LMWH/warfarin/DOACs)
- Corticosteroids (short-term during acute hemolytic crises - debated benefit)
- Immunosuppression (for AA component - anti-thymocyte globulin + cyclosporine)
3. Curative Therapy
- Allogeneic hematopoietic stem cell transplantation (HSCT) - the only cure
- Reserved for: aplastic anemia with PNH clone, transformation to AML/MDS, recurrent life-threatening thrombosis despite complement inhibition, young patients with HLA-matched sibling
Key Concepts Summary
PIGA mutation (somatic, X-linked)
↓
GPI anchor deficiency
↓
Loss of CD55, CD59, C8-BP on ALL blood cell lineages
↓
┌───────────────────────────────────────────────┐
│ Uncontrolled complement activation on RBCs │
│ MAC (C5b-9) assembly → Intravascular hemolysis│
│ Hemoglobinuria + Hemosiderinuria │
└───────────────────────────────────────────────┘
↓
┌──────────────────────────────────────────────┐
│ Free Hb → NO scavenging → smooth muscle │
│ dystonia (dysphagia, abdo pain, erectile dys)│
└──────────────────────────────────────────────┘
↓
┌──────────────────────────────────────────────┐
│ CD59-deficient platelets → Thrombosis │
│ (Budd-Chiari, cerebral, portal vein) │
│ → LEADING CAUSE OF DEATH │
└──────────────────────────────────────────────┘
DIAGNOSIS: Flow cytometry (CD55, CD59, FLAER)
Coombs NEGATIVE
TREATMENT: Eculizumab/Ravulizumab (anti-C5)
HSCT (only cure)
Robbins, Cotran & Kumar Pathologic Basis of Disease | Harrison's Principles of Internal Medicine, 22nd Ed. | Goldman-Cecil Medicine, International Ed. | Henry's Clinical Diagnosis and Management by Laboratory Methods
Classify myelodysplastic syndrome and mention diagnostic criteria, for 10 marks
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Reading File
I now have all the content needed for a comprehensive 10-mark answer. Let me compile it.
Myelodysplastic Syndrome (MDS): Classification and Diagnostic Criteria
Definition
Myelodysplastic Syndromes (MDS) are a heterogeneous group of clonal hematopoietic stem cell disorders characterized by:
- Ineffective hematopoiesis - dysplastic (morphologically abnormal) maturation in one or more blood cell lineages
- Peripheral blood cytopenias (despite a usually normo- or hypercellular bone marrow)
- Variable risk of transformation to Acute Myeloid Leukemia (AML)
MDS is primarily a disease of older adults (median age ~70 years) and represents a pre-leukemic state in many cases.
Pathogenesis (Brief)
A somatic mutation occurs in a pluripotent hematopoietic stem cell → clonal proliferation dominates the marrow. Bone marrow is usually normo- or hypercellular, yet excessive apoptosis of developing cells causes peripheral cytopenias ("ineffective hematopoiesis"). As the disease progresses toward AML, intramedullary apoptosis decreases and hematopoietic differentiation becomes arrested.
Commonly mutated genes (>40 identified):
- Spliceosome components: SF3B1 (20-25%), SRSF2 (10-15%), U2AF1 (10%) - strongly associated with ring sideroblasts when SF3B1 is mutated
- DNA methylation/epigenetics: TET2 (20-25%), DNMT3A (10-15%), ASXL1 (15-20%)
- Transcription factors: RUNX1 (10-15%), ETV6 (3%)
- Tumor suppressors: TP53 (5-10%) - poor prognosis
- RAS pathway: NRAS, KRAS (≤10%)
Chromosomal abnormalities present in ~50% of de novo MDS and >80% of therapy-related MDS.
WHO Classification of MDS
The WHO 5th Edition (2022) classifies MDS based on: dysplastic lineages, blast counts, ring sideroblasts, Auer rods, and cytogenetic/molecular findings. Note: the WHO 2022 renames MDS subtypes as "Myelodysplastic Neoplasms" to reflect their clonal nature.
WHO MDS Subtypes (Goldman-Cecil Medicine, Table 162)
1. MDS with Single Lineage Dysplasia (MDS-SLD)
| Feature | Criteria |
|---|---|
| Dysplastic lineages | 1 lineage only (>10% of cells in that lineage) |
| Peripheral blood blasts | <1% |
| Bone marrow blasts | <5% |
| Ring sideroblasts | <15% of erythroid precursors |
| Cytopenias | 1-2 |
| Auer rods | Absent |
Comment: Includes cases with cytokine gene mutations, but marrow dysplasia limited to one lineage.
2. MDS with Ring Sideroblasts (MDS-RS)
| Feature | Criteria |
|---|---|
| Dysplasia | Erythroid lineage (>10% of erythroid precursors) |
| Ring sideroblasts | ≥15% of erythroid precursors (or ≥5% if SF3B1 mutation present) |
| Peripheral blood blasts | None |
| Bone marrow blasts | <5% |
| Anemia | Normocytic/macrocytic |
Subtypes:
- MDS-RS-SLD - single lineage dysplasia
- MDS-RS-MLD - multilineage dysplasia
Ring sideroblast: An erythroid precursor with iron granules (visible on Perls' Prussian blue) surrounding at least one-third of the nucleus in a perinuclear ring pattern - reflects abnormal mitochondrial iron accumulation.
3. MDS with Multilineage Dysplasia (MDS-MLD)
| Feature | Criteria |
|---|---|
| Dysplastic lineages | 2 or more lineages (>10% of cells in each affected lineage) |
| Peripheral blood blasts | <1% |
| Bone marrow blasts | <5% |
| Auer rods | Absent |
| Cytopenias | One or more |
Note: If ≥15% ring sideroblasts (or ≥5% with SF3B1 mutation) → reclassify as MDS-RS-MLD.
4. MDS with Excess Blasts-1 (MDS-EB-1)
| Feature | Criteria |
|---|---|
| Dysplasia | 1 or more lineages |
| Peripheral blood blasts | 2-4% |
| Bone marrow blasts | 5-9% |
| Auer rods | Absent |
| Cytopenias | One or more |
Note: 2-4% blasts in PB → automatically MDS-EB-1 regardless of marrow blast count.
5. MDS with Excess Blasts-2 (MDS-EB-2)
| Feature | Criteria |
|---|---|
| Dysplasia | 1 or more lineages |
| Peripheral blood blasts | 5-19% |
| Bone marrow blasts | 10-19% |
| Auer rods | Presence of Auer rods at ANY blast count <20% → MDS-EB-2 |
| Cytopenias | One or more |
Higher risk of AML transformation; 20% marrow blasts = AML threshold (WHO 2022: 10-19% may be classified as MDS/AML overlap).
6. MDS with Isolated del(5q) - "5q- Syndrome"
| Feature | Criteria |
|---|---|
| Cytogenetics | Isolated deletion of chromosome 5q (may have one additional cytogenetic abnormality other than monosomy 7) |
| Megakaryocytes | Increased; small, hypolobated or non-lobated nuclei (pathognomonic) |
| Granulocytic dysplasia | Minimal or absent |
| Peripheral blood blasts | None or rare (<1%) |
| Bone marrow blasts | <5% |
| Anemia | Macrocytic |
Special significance:
- Predominantly affects middle-aged women
- Lenalidomide is specifically effective for this subtype
- Auer rods must be absent; del(5q) + monosomy 7 = excluded from this category (worse prognosis)
7. MDS, Unclassifiable (MDS-U)
Cases that do not fit into the above categories; includes:
- Persistent cytopenias with <10% dysplastic cells but a characteristic MDS-associated cytogenetic abnormality
- Cases with 1% peripheral blood blasts on two occasions
- Cases with single lineage dysplasia + pancytopenia
Additional WHO 2022 Categories
| Category | Key Feature |
|---|---|
| MDS with low blasts and SF3B1 mutation | SF3B1 mutation + <15% ring sideroblasts; low blast count |
| MDS with low blasts | Low blast count without specific cytogenetic or molecular marker |
| MDS with biallelic TP53 inactivation | Biallelic TP53 mutation; complex karyotype; very poor prognosis |
| Hypoplastic MDS | Hypocellular marrow; overlaps with aplastic anemia |
| Therapy-related MDS (t-MDS) | Following cytotoxic therapy/radiotherapy; often TP53 or complex karyotype |
| MDS/AML | 10-19% blasts (WHO 2022 overlap category) |
Diagnostic Criteria
Minimal Diagnostic Criteria (Goldman-Cecil Medicine)
A diagnosis of MDS requires one or more cytopenias PLUS at least one of the following:
| Criterion | Threshold |
|---|---|
| Marrow myeloblasts | 5-19% |
| Dysplastic cells in any lineage | >10% of cells in a given hematopoietic lineage |
| Characteristic cytogenetic abnormality | Acquired clonal chromosomal anomaly consistent with MDS diagnosis |
After exclusion of other causes of dysplasia (B12/folate deficiency, drug effects, infections)
Step-by-Step Diagnostic Work-Up
1. Clinical Suspicion
- Older patient with unexplained cytopenias (especially macrocytic anemia not explained by B12/folate deficiency, drugs, or alcohol)
- Fatigue, dyspnea, recurrent infections, mucosal bleeding
2. Peripheral Blood Examination
Cytopenia thresholds:
- Hb <10 g/dL (or <13 g/dL in males)
- ANC <1.8 × 10⁹/L
- Platelets <100 × 10⁹/L
Dysplastic morphology on peripheral smear:
MDS dysplastic morphology: (A) Multinucleated erythroid precursor; (B) Megaloblastoid erythroid maturation with open chromatin; (C) Ring sideroblast - Perls' Prussian blue; (D) Hypogranular neutrophils; (E) Pseudo-Pelger-Huet (bilobed/hypolobated) neutrophils; (F) Megakaryocyte with separated nuclear lobes ("pawn ball") - Goldman-Cecil Medicine
| Cell Line | Dysplastic Features in Peripheral Blood |
|---|---|
| Erythroid | Poikilocytosis, anisocytosis, macro-ovalocytes, basophilic stippling, nucleated RBCs |
| Myeloid (Granulocytes) | Hypogranular neutrophils, pseudo-Pelger-Huet anomaly (bilobed/non-segmented nucleus), hypersegmented forms |
| Megakaryocytes/Platelets | Giant platelets, platelet hypogranularity, micromegakaryocytes in marrow |
3. Bone Marrow Examination (Essential)
Both aspirate AND trephine biopsy required:
| Bone Marrow Finding | Significance |
|---|---|
| Cellularity | Usually normo- or hypercellular (hypocellular in ~15%) |
| Erythroid dysplasia | Multinucleated erythroblasts, megaloblastoid changes, nuclear budding, karyorrhexis |
| Ring sideroblasts | Perls' Prussian blue - perinuclear iron granules (≥1/3 nuclear circumference); ≥15% of erythroid precursors (or ≥5% + SF3B1) |
| Myeloid dysplasia | Left-shift, hypogranularity, pseudo-Pelger-Huet, Auer rods |
| Megakaryocyte dysplasia | Micromegakaryocytes, hypolobated nuclei, widely separated nuclear lobes |
| Blast percentage | 5-19% = MDS-EB; ≥20% = AML |
| Reticulin fibrosis | Mild increase common; severe/collagen fibrosis rare |
4. Cytogenetics (Essential)
Metaphase karyotype on bone marrow aspirate is mandatory (at least 20 metaphases). FISH used only if karyotyping fails.
Common MDS-associated cytogenetic abnormalities:
| Abnormality | Association |
|---|---|
| del(5q) | MDS-5q syndrome; good prognosis; responds to lenalidomide |
| -7 or del(7q) | Poor prognosis |
| +8 (trisomy 8) | Common; intermediate prognosis |
| del(20q) | Relatively favorable |
| -Y | Relatively favorable |
| -5 or del(5q) + other | Poor prognosis (therapy-related) |
| Complex karyotype (≥3 abnormalities) | Very poor prognosis; often TP53-mutated |
| Monosomal karyotype | Worst prognosis |
5. Molecular/Genetic Testing (Next-Generation Sequencing)
Not strictly required for diagnosis but provides adjunctive information:
- SF3B1 mutation → confirms MDS-RS even with <15% ring sideroblasts (≥5% sufficient)
- TP53 biallelic mutations → distinct entity; very poor prognosis
- RUNX1 → thrombocytopenic prodrome
- GATA2 → mycobacterial infections, monocytopenia
6. Flow Cytometry
- Useful for diagnosing PNH clone (in hypoplastic MDS/AA overlap)
- Immunophenotyping can support diagnosis but should not be used to count blast percentage
Prognostic Scoring: IPSS-R
The Revised International Prognostic Scoring System (IPSS-R) stratifies MDS into 5 risk groups based on:
- Cytogenetics (5 categories: very good → very poor)
- Marrow blast %
- Hemoglobin
- Platelet count
- ANC
| Risk Category | Median Survival |
|---|---|
| Very Low | 8.8 years |
| Low | 5.3 years |
| Intermediate | 3.0 years |
| High | 1.6 years |
| Very High | 0.8 years |
Higher-risk MDS (IPSS-R High/Very High): Immediate therapy; life expectancy <2 years without treatment.
Differential Diagnosis
Other causes of cytopenias and dysplasia that must be excluded before diagnosing MDS:
- B12 / folate deficiency (megaloblastic anemia)
- Copper deficiency (especially if ring sideroblasts)
- Iron deficiency
- HIV infection
- Drug effects (cytotoxic chemotherapy, methotrexate, azathioprine)
- Heavy alcohol use
- Congenital sideroblastic anemias (ALAS2 mutations)
- Aplastic anemia
- T-cell large granular lymphocyte (T-LGL) leukemia
Summary Table
| MDS Subtype | Key Features | BM Blasts | PB Blasts |
|---|---|---|---|
| MDS-SLD | 1 lineage dysplastic | <5% | <1% |
| MDS-RS (SLD/MLD) | ≥15% ring sideroblasts (or ≥5% + SF3B1) | <5% | None |
| MDS-MLD | ≥2 lineages dysplastic | <5% | <1% |
| MDS-EB-1 | Excess blasts, no Auer rods | 5-9% | 2-4% |
| MDS-EB-2 | Excess blasts ± Auer rods | 10-19% | 5-19% |
| MDS with del(5q) | Isolated del(5q), hypolobated megakaryocytes | <5% | None/rare |
Goldman-Cecil Medicine, International Ed. | Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.