Dcld
Reading File
Finding Sources
Searching the Web
DCLD medical abbreviation decompensated chronic liver disease
Asking for Preferences
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Here is a structured overview of the causes and pathophysiology of Decompensated Chronic Liver Disease (DCLD):
DCLD - Causes & Pathophysiology
Definition
Decompensated chronic liver disease (DCLD) refers to the stage where cirrhosis - the end-stage of chronic liver injury - leads to clinical complications that the liver can no longer compensate for. These hallmark decompensating events include ascites, variceal hemorrhage, hepatic encephalopathy, jaundice, and coagulopathy.
Causes (Etiology)
Any condition causing chronic hepatic inflammation and injury can progress to cirrhosis and eventual decompensation. The major categories are:
| Category | Examples |
|---|---|
| Viral | Chronic hepatitis B (HBV), chronic hepatitis C (HCV) |
| Toxic/Metabolic | Alcoholic liver disease, non-alcoholic steatohepatitis (NASH/MAFLD) |
| Metabolic/Genetic | Hemochromatosis, Wilson disease, alpha-1 antitrypsin deficiency, cystic fibrosis |
| Autoimmune/Cholestatic | Autoimmune hepatitis, primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC) |
| Vascular | Budd-Chiari syndrome, congestive hepatopathy |
| Parasitic | Schistosomiasis (major worldwide cause) |
| Drugs | Methotrexate, amiodarone, other hepatotoxins |
| Other | Sarcoidosis, neonatal liver disease, cryptogenic cirrhosis |
In the United States, the leading causes are HCV, NASH, and alcoholic liver disease (Yamada's Textbook of Gastroenterology, p. 2085). Globally, viral hepatitis and parasitic infection (especially schistosomiasis) dominate.
Pathophysiology
DCLD is driven by two interconnected mechanisms: progressive hepatic fibrosis and portal hypertension with its downstream consequences.
1. Hepatic Fibrosis and Stellate Cell Activation
In response to chronic liver injury, the liver undergoes a maladaptive wound-healing response:
- Hepatocyte injury (from viruses, alcohol, fat, toxins) triggers Kupffer cell activation and inflammation
- Inflammatory mediators (TNF-alpha, IL-6, TGF-beta, PDGF) activate hepatic stellate cells (HSCs) - the key cellular effectors of fibrosis
- Activated HSCs transform into myofibroblast-like cells and produce excess extracellular matrix (ECM) - predominantly collagen types I and III
- Accumulation of ECM distorts hepatic architecture, forming regenerative nodules surrounded by fibrous bands - this is cirrhosis
Key profibrogenic mediators include TGF-beta (strongest profibrotic cytokine), PDGF, endothelin-1, and angiotensin II. (Yamada's Textbook of Gastroenterology, p. 2078)
2. Portal Hypertension and the Hyperdynamic Circulation
Cirrhosis raises portal pressure through two mechanisms:
- Anatomical: fibrosis and regenerative nodules block intrahepatic blood flow
- Functional: intrahepatic vasoconstriction due to decreased nitric oxide (NO) production
At the same time, extrahepatic splanchnic vasodilation occurs (paradoxically due to excess NO in the systemic circulation), which:
- Decreases effective arterial blood volume
- Activates the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS)
- Raises plasma ADH
This creates a hyperdynamic circulatory state (high cardiac output, low SVR) that sustains portal hypertension and drives all major complications. (Goldman-Cecil Medicine, p. ch. 139)
3. Major Pathophysiological Cascades Leading to Decompensation
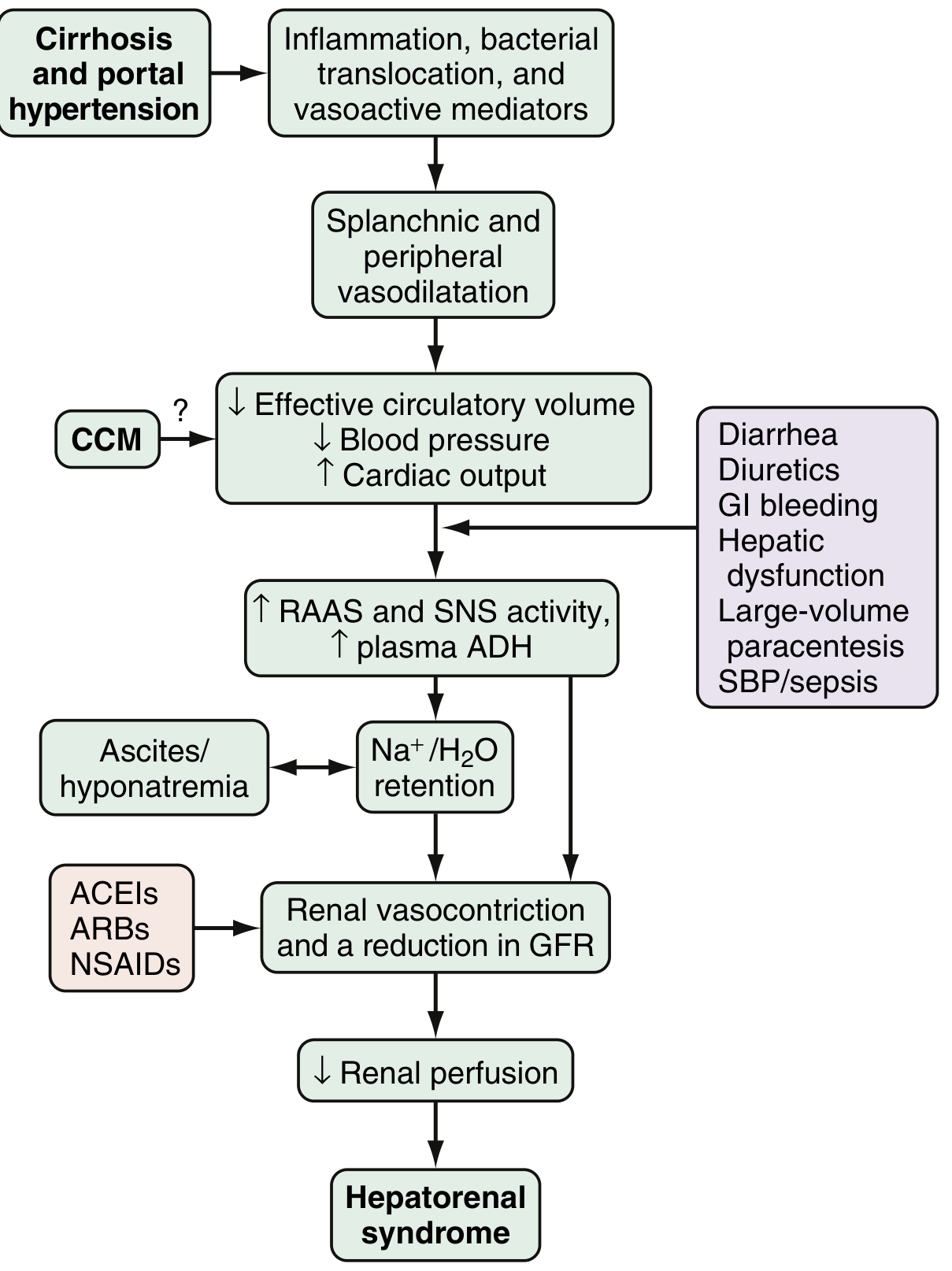
The diagram below (from Sleisenger & Fordtran) illustrates how cirrhosis + portal hypertension leads to each decompensating event:
A. Ascites
- Sinusoidal hypertension (HVPG > 12 mmHg threshold) + sodium/water retention (via RAAS) = fluid leaking into the peritoneal cavity
- With progression: refractory ascites due to worsening RAAS/SNS activation
B. Variceal Hemorrhage
- HVPG > 10-12 mmHg triggers portosystemic collateral formation (esophageal and gastric varices)
- Hyperdynamic state dilates varices further until they rupture
- Variceal wall tension = (variceal radius x intravariceal pressure) / wall thickness (LaPlace's law)
C. Hepatorenal Syndrome (HRS)
- Progressive splanchnic vasodilation is not fully compensated --> decreased renal perfusion
- RAAS/SNS activation causes renal vasoconstriction despite global fluid overload (the "paradox" of HRS)
- Triggers include: diarrhea, diuretics, GI bleeding, spontaneous bacterial peritonitis (SBP), large-volume paracentesis
D. Spontaneous Bacterial Peritonitis (SBP)
- Bacterial translocation from gut lumen to mesenteric lymph nodes (driven by disrupted gut barrier and altered microbiome)
- Impaired Kupffer cell function + portosystemic shunting allows bacteremia to persist
- Colonizes ascitic fluid with low opsonic activity
E. Hepatic Encephalopathy (HE)
- Ammonia is the central toxin: impaired hepatic ureagenesis + portosystemic shunting allows ammonia to reach the brain
- Ammonia causes astrocyte swelling and cerebral dysfunction
- Additional contributors: inflammatory mediators, gut microbiome dysbiosis, GABA system dysregulation
- Portosystemic shunting (bypassing liver) is a prerequisite alongside liver insufficiency
F. Jaundice and Coagulopathy
- Solely from hepatocellular insufficiency: reduced synthetic function (clotting factors I, II, V, VII, IX, X), reduced bilirubin conjugation and excretion
Summary Diagram of Pathogenesis
Chronic Liver Injury (Viral / Alcohol / NASH / Metabolic / Immune)
↓
Stellate cell activation → ECM/collagen deposition → CIRRHOSIS
↓
Architectural distortion + ↓ intrahepatic NO
↓
Portal Hypertension (HVPG ↑)
↓
Splanchnic vasodilation (↑ systemic NO)
↓
↓ Effective circulating volume → RAAS/SNS/ADH activation
↓
Ascites ─── Varices ─── HRS ─── SBP ─── HE ─── Jaundice/Coagulopathy
[= DECOMPENSATION]
Sources: Yamada's Textbook of Gastroenterology 7th Ed.; Goldman-Cecil Medicine; Sleisenger & Fordtran's GI & Liver Disease; Harrison's Principles of Internal Medicine 22E; Tietz Textbook of Laboratory Medicine 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.