Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND mechanism pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive mechanistic content and two excellent images. I have enough to write a thorough, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS) — Mechanism
ARDS is a syndrome of non-cardiogenic pulmonary edema caused by diffuse alveolar damage (DAD), resulting in severe hypoxemia, reduced lung compliance, and bilateral infiltrates. It is defined by the Berlin criteria: acute onset (<1 week), bilateral opacities on imaging, PaO₂/FiO₂ ratio <300 mmHg with ≥5 cmH₂O PEEP, and absence of cardiogenic cause.
1. Initiating Insults — Direct vs. Indirect
ARDS is triggered by either direct (pulmonary) or indirect (extrapulmonary) injury:
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Pancreatitis |
| Pulmonary contusion | Massive transfusion |
| Inhalation injury | Trauma / burns |
| Near-drowning | Drug overdose |
2. Core Pathophysiological Event — Alveolar-Capillary Barrier Breakdown
The central mechanism is loss of alveolar-capillary membrane integrity, involving both:
- Pulmonary microvascular endothelium — loss of endothelial barrier function is both necessary and sufficient for ARDS development.
- Alveolar epithelium — damage to type I and type II pneumocytes disrupts barrier integrity AND prevents alveolar fluid clearance. Epithelial cell death occurs via necrosis, apoptosis, coagulation-mediated mechanisms, and mechanical stretch.
The result is flooding of the alveolar space with protein-rich edema fluid, plasma proteins, erythrocytes, and inflammatory cells.
3. Neutrophil-Mediated Injury (Central Effector)
One of the histological hallmarks of ARDS is accumulation of neutrophils in the pulmonary microvasculature and alveolar spaces.
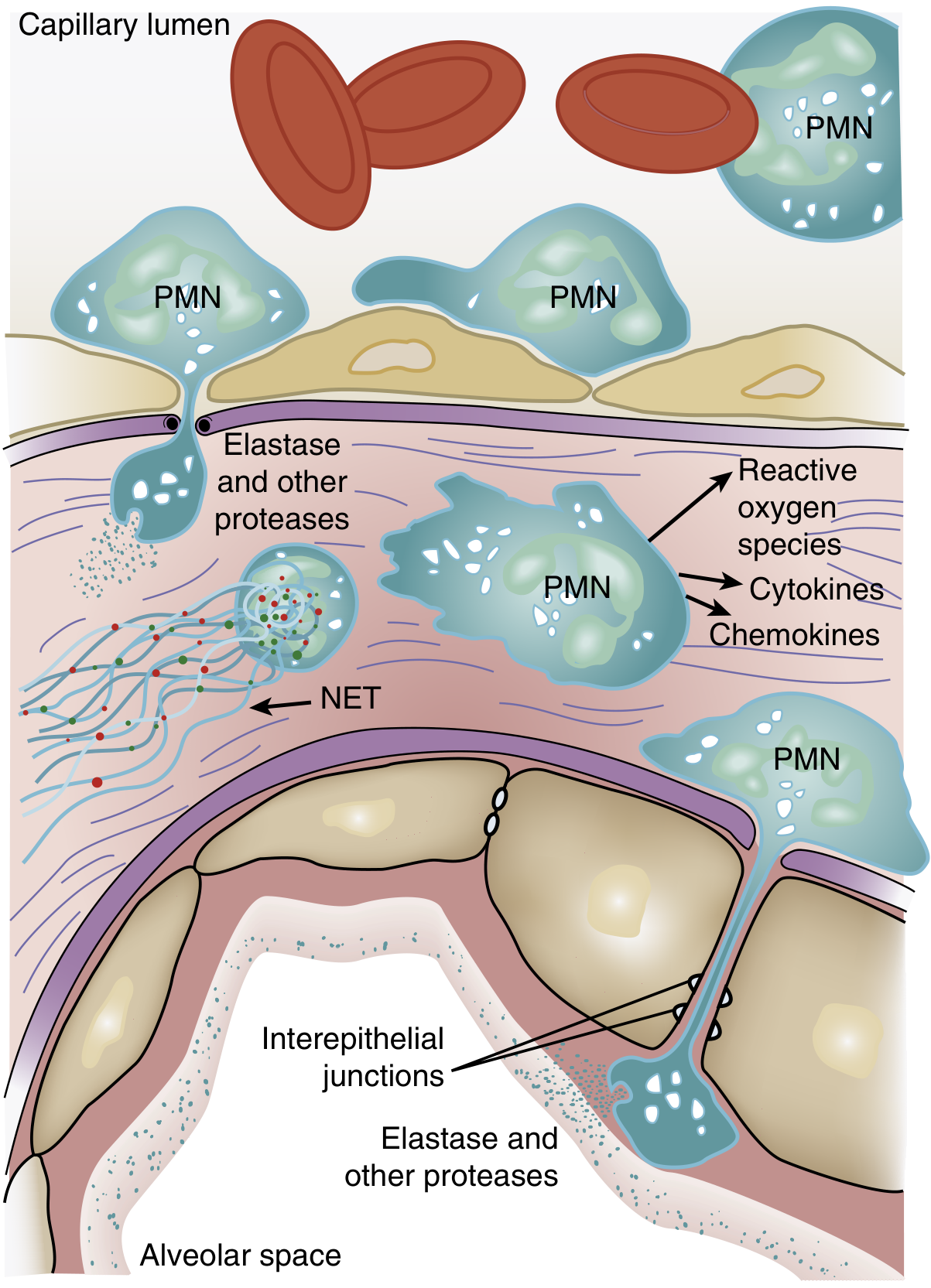
Mechanism of neutrophil sequestration:
- The average pulmonary capillary is narrower than the average neutrophil diameter; neutrophils must deform to pass through.
- Activated neutrophils become "stiff" (actin cytoskeleton changes) and cannot negotiate capillary segments → they are sequestered in the pulmonary microcirculation. This causes a transient leukopenia — often one of the earliest signs of ARDS.
- Sequestered neutrophils promote endothelial barrier breakdown, facilitating further transmigration into the interstitium and alveolar space.
Cytotoxic arsenal released by neutrophils:
- Reactive oxygen species (ROS) — oxidative damage to membranes and proteins
- Neutrophil elastase (NE) — degrades epithelial/endothelial cadherins (components of adherens junctions), predisposing to alveolar flooding; also degrades surfactant protein A
- Matrix metalloproteinases — proteolytic degradation of extracellular matrix
- Cationic peptides (defensins) — direct cytotoxicity
- Eicosanoids — amplify inflammation
- Cytokines (TNF-α, IL-1β) — amplify the inflammatory cascade
Neutrophil extracellular traps (NETs):
NETs are web-like structures of DNA, histones, and antimicrobial peptides (myeloperoxidase, elastase, cathepsin G) released by neutrophils. In sepsis, large-scale NET formation causes endothelial damage and thrombus formation. In animal models, NET formation in the lung is accompanied by severe structural lung destruction; DNase treatment or NE inhibition attenuates lung injury and lowers IL-6 and TNF levels.
Note: ARDS can occur in profoundly neutropenic patients, implying neutrophil-independent pathways also exist — alveolar macrophages may serve as alternative injury mediators in this setting.
4. The Cytokine Storm and Inflammatory Amplification
Multiple pro-inflammatory mediators amplify and sustain lung injury:
- TNF-α and IL-1β — early mediators released by macrophages and activated neutrophils; upregulate adhesion molecules, activate endothelium, and recruit more neutrophils
- IL-8 — a major chemokine driving neutrophil recruitment via CXCR1/CXCR2 receptors
- Phosphatidylinositol 3-kinase-γ signaling — activated by IL-8 and bacterial peptides in neutrophils, amplifies cytokine production and neutrophil accumulation
- Platelet-activating factor, prostaglandins, leukotrienes — further increase vascular permeability
- Reactive oxygen and nitrogen species — from both neutrophils and activated macrophages
In pancreatitis-associated ARDS, activated pancreatic enzymes (phospholipase A₂, elastase, lipase) play an additional direct role: phospholipase A₂ enzymatically degrades surfactant and increases vascular permeability.
5. Surfactant Dysfunction
Type II pneumocytes produce surfactant, which reduces surface tension and prevents alveolar collapse. In ARDS:
- Surfactant production is decreased due to type II pneumocyte injury
- The ratio of large (active) to small (inactive) surfactant aggregates is diminished
- Plasma proteins leaking into the alveolus interfere with surfactant function
- Neutrophil elastase degrades surfactant protein A
The result: alveoli are prone to collapse at end-expiration, dramatically reducing functional residual capacity (FRC) and worsening V/Q mismatch and intrapulmonary shunting.
6. Coagulation and Fibrin Deposition
Widespread endothelial injury triggers the coagulation cascade within lung microvessels:
- Fibrin thrombi form in pulmonary capillaries → ischemic injury to alveolar cells
- Fibrin exudate in alveolar spaces → forms the characteristic hyaline membranes seen histologically
- Impaired fibrinolysis perpetuates microvascular obstruction, contributing to dead-space physiology (high V/Q units)
7. The Three Phases of ARDS
Phase 1 — Exudative (Days 1–7)
- Alveolar-capillary barrier breakdown
- Flooding with protein-rich edema fluid
- Hyaline membrane formation (fibrin + cellular debris)
- Neutrophil infiltration, diffuse alveolar damage
- Type I pneumocyte necrosis → denuded basement membrane
- Clinically: acute onset severe hypoxemia, bilateral infiltrates
Phase 2 — Proliferative/Fibroproliferative (Days 7–21)
- Type II pneumocyte proliferation to re-epithelialize denuded areas
- Fibroblast migration and proliferation → early collagen deposition
- Organizing pneumonia pattern
- Resolution of edema may begin in survivors
- Continued cytokine-driven inflammation in non-resolving cases
Phase 3 — Fibrotic (>3 weeks, in some patients)
- Extensive collagen deposition and obliteration of normal alveolar architecture
- Pulmonary hypertension from vascular remodeling
- Severely reduced compliance
- Associated with high mortality and long-term impaired quality of life
8. Physiological Consequences
| Mechanism | Physiological Effect |
|---|---|
| Alveolar flooding | ↓ FRC, ↓ compliance |
| Alveolar collapse (atelectasis) | Intrapulmonary shunt (V/Q = 0) → refractory hypoxemia |
| Microvascular obstruction | Dead-space ventilation (V/Q = ∞) → hypercapnia in late disease |
| Surfactant loss | Alveolar instability, recruitment-derecruitment with each breath |
| Pulmonary hypertension | Right ventricular strain, ↓ cardiac output |
The heterogeneous nature of ARDS — with some alveoli flooded, others collapsed, and others relatively normal — creates the "baby lung" concept: only a small volume of lung participates in gas exchange, making standard tidal volumes potentially injurious (volutrauma/barotrauma).
Summary Pathway
Initiating insult (sepsis, pneumonia, aspiration, etc.)
↓
Systemic/local inflammatory activation
↓
Neutrophil sequestration in pulmonary microcirculation
↓
Neutrophil transmigration → release of ROS, elastase, proteases, NETs, cytokines
↓
Alveolar-capillary barrier breakdown
(endothelial + epithelial injury)
↓
Protein-rich edema floods alveoli + surfactant dysfunction
↓
Hyaline membrane formation + coagulation activation
↓
↓ FRC, ↓ compliance, intrapulmonary shunt, V/Q mismatch
↓
Severe refractory hypoxemia → ARDS
Sources: Murray & Nadel's Textbook of Respiratory Medicine (Chapter 134); Goldman-Cecil Medicine (Chapter 90, 91)
This is a shared conversation. Sign in to Orris to start your own chat.