Treatment of Bernard souiler syndrome
Reading File
Finding Sources
Searching PubMed
"Bernard-Soulier syndrome" AND treatment
Reading File
Reading File
Reading Article
PMID: 39191409
Reading File
Reading Article
PMID: 41746364
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Bernard-Soulier Syndrome (BSS) - Treatment
Background
BSS is a rare autosomal recessive inherited platelet function disorder caused by deficiency or dysfunction of the GPIb-IX-V complex (mutations in GPIBA, GPIBB, or GP9). This complex normally binds von Willebrand factor (vWF), mediating platelet adhesion to subendothelium. Without it, primary hemostatic plug formation fails. The result is macrothrombocytopenia with mucocutaneous bleeding of varying severity.
Treatment Overview
Treatment is triggered by:
- Active bleeding episodes
- Pre-procedural/surgical prophylaxis
- Delivery in pregnant women (with significant bleeding history)
1. Platelet Transfusion (Mainstay of Treatment)
- HLA-matched, single-donor platelets are the primary treatment for significant bleeding.
- Matching is important because repeated transfusions lead to alloimmunization against platelet-specific antigens and HLA antigens, causing transfusion refractoriness.
- Limiting unnecessary transfusions is therefore critical - they should be reserved for active bleeding or high-risk procedures.
- In the obstetric setting, routine prophylactic platelet transfusion before delivery is controversial due to the risk of alloimmune thrombocytopenia (AIT) - reserved only for those with severe bleeding history.
- Harrison's Principles of Internal Medicine 22E (2025), p. 969; Goldman-Cecil Medicine, p. 1823
2. Recombinant Factor VIIa (rFVIIa / Eptacog alfa)
- rFVIIa is FDA approved for use in both Glanzmann thrombasthenia and Bernard-Soulier syndrome.
- Indicated especially when patients are alloimmunized and refractory to platelet transfusion.
- Mechanism: enhances thrombin generation at the site of vascular injury, thereby boosting platelet recruitment independently of the GPIb-IX-V complex.
- A 2026 case report (Okamoto et al., Ann Hematol) described successful hemostasis with rFVIIa in a BSS patient refractory to HLA-matched platelets following trauma, with no thromboembolic complications. [PMID: 41746364]
- Dosing: typically follows the regimen used in Glanzmann thrombasthenia surgical settings.
- Harrison's, p. 969; Goldman-Cecil, p. 1824
3. DDAVP (Desmopressin)
- Useful for mild to moderate bleeding in BSS, particularly as an adjunct.
- Mechanism: raises plasma vWF and factor VIII levels; may also have direct platelet effects.
- Administered IV at 0.3 mcg/kg.
- Caution: can cause fluid retention and hyponatremia - monitor electrolytes.
- Not sufficient as monotherapy for severe bleeding.
- Goldman-Cecil Medicine, p. 1823-1824; Creasy & Resnik's Maternal-Fetal Medicine
4. Antifibrinolytic Therapy
Especially effective for mucosal bleeding (epistaxis, menorrhagia, oral/dental bleeding):
| Drug | Dose |
|---|---|
| Tranexamic acid | 15-25 mg/kg orally TID; or mouth rinse for oral bleeding |
| Epsilon-aminocaproic acid (EACA) | 5 g orally, then 1-1.25 g/hr orally for 8 hours (max 30 g/day) |
- Used alone or in combination with DDAVP or platelet transfusion.
- Particularly valuable in tissues of high fibrinolytic activity (nose, mouth, throat, extraocular tissue).
- Goldman-Cecil Medicine, p. 1824; Scott-Brown's Otorhinolaryngology
5. Oral Contraceptives
- Used in women with heavy menstrual bleeding (menorrhagia) as supplementary treatment.
- Reduces menstrual blood loss by suppressing endometrial proliferation.
- Kaya Z, Semin Thromb Hemost, 2025 [PMID: 39191409]
6. Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)
- Reserved for severe, refractory cases unresponsive to other therapies.
- The only potential curative option currently available.
- Risk-benefit must be carefully considered given transplant-related morbidity.
- Kaya Z, Semin Thromb Hemost, 2025 [PMID: 39191409]
Special Situations
Obstetric / Peripartum Management
- Postpartum hemorrhage occurs in ~60% of BSS pregnancies; ~50% require blood transfusion (Creasy & Resnik's Maternal-Fetal Medicine).
- HLA-matched platelets preferred to reduce AIT risk.
- Cesarean delivery should be reserved for obstetric indications only.
- Neuraxial anesthesia should be avoided - normal hemostasis is not guaranteed even after prophylactic platelet transfusion.
- DDAVP, tranexamic acid, and rFVIIa are adjuncts for peripartum bleeding control.
Avoid Antiplatelet Drugs
- NSAIDs, aspirin, and other antiplatelet agents must be strictly avoided as they worsen platelet dysfunction.
Treatment Summary Table
| Situation | First Line | Second/Adjunct |
|---|---|---|
| Severe/active bleeding | HLA-matched platelet transfusion | rFVIIa |
| Alloimmunized / transfusion-refractory | rFVIIa | - |
| Mild-moderate bleeding | DDAVP | Antifibrinolytics |
| Mucosal bleeding | Antifibrinolytics (tranexamic acid) | DDAVP |
| Menorrhagia | Oral contraceptives | Tranexamic acid |
| Refractory severe disease | Allogeneic HSCT | - |
| Pre-procedure prophylaxis | HLA-matched platelets (if severe history) | rFVIIa |
Recent evidence note: A 2025 comprehensive review (Kaya, Semin Thromb Hemost 2025, PMID 39191409) confirmed platelet transfusion remains the cornerstone, with rFVIIa recommended in alloimmunized patients and HSCT for refractory cases - consistent with current textbook recommendations. A 2026 case report further supports rFVIIa as an effective rescue option in transfusion-refractory BSS.
Treatment and cause of premature greying of hair
Finding Sources
Searching PubMed
"premature hair graying" AND (treatment OR cause OR pathogenesis)
Reading File
Reading Article
PMID: 39697103
Finding Sources
Reading File
Reading File
Finding Sources
Searching the Web
premature hair greying causes treatment nutritional deficiency 2024 2025
Reading File
Reading File
I now have comprehensive material from multiple dermatology textbooks plus the 2025 PubMed review. Let me compile the full answer.
Premature Greying of Hair (Canities Praecox)
Definition
Premature greying is defined as onset of hair greying before age 20 in White/Asian individuals and before age 30 in Black individuals. Physiologic greying typically begins at the temples and progresses - beard follows scalp, body hair greys last.
Pathophysiology
Hair colour is produced by follicular melanocytes in the hair bulb matrix during the anagen phase. Greying results from:
- Depletion of melanocyte stem cells in the hair follicle bulge region - reduced migration replenishes fewer active melanocytes each hair cycle.
- Loss of tyrosinase activity within bulbar melanocytes - this enzyme is rate-limiting for melanin synthesis.
- Reactive oxygen species (ROS) / H₂O₂ accumulation - oxidative damage to melanocyte DNA (nuclear and mitochondrial) is a key mechanism. Catalase activity declines with age; accumulated H₂O₂ bleaches hair from within and impairs methionine sulfoxide repair.
- Reduction in melanosomes - fewer and smaller melanosomes transferred to cortical keratinocytes.
- Andrews' Diseases of the Skin; Fitzpatrick's Dermatology; Dermatology 2-Volume Set 5e
Causes
A. Genetic / Hereditary
- Familial (autosomal dominant) - the most common cause; positive family history is the strongest predictor.
- Accounts for early greying that is otherwise unexplained.
- IRF4, MCIR, and other pigmentation gene variants influence onset.
B. Nutritional Deficiencies
| Deficiency | Notes |
|---|---|
| Vitamin B12 | Causes whitening/greying; seen with metformin use, antacids, H. pylori eradication therapy, pernicious anaemia |
| Iron deficiency | "Canities segmentata sideropenica" - banding/greying that responds completely to iron supplementation |
| Copper | Cofactor for tyrosinase; deficiency impairs melanin synthesis |
| Vitamin D | Emerging evidence of association |
| Ferritin | Low stores correlate with premature greying |
| Protein (kwashiorkor) | Causes flag sign - alternating light/dark banding |
C. Endocrine / Autoimmune Disorders
- Vitiligo - most common acquired cause; anti-melanocyte autoimmunity destroys follicular melanocytes (can occur without visible skin lesions)
- Thyroid disorders (hypo- and hyperthyroidism)
- Alopecia areata - "canities subita" (sudden whitening) occurs when all pigmented hairs fall out, leaving only white ones
- Pernicious anaemia (B12 deficiency via intrinsic factor loss)
D. Environmental / Lifestyle
- Smoking - well-established independent risk factor (oxidative stress)
- Chronic psychological stress - activates sympathetic nervous system, depletes melanocyte stem cell pool (documented in animal models and human studies)
- Radiation exposure
- Oxidative stress from UV, pollution, or nutritional antioxidant deficiency
E. Drugs / Medications
| Drug | Effect |
|---|---|
| Chloroquine / hydroxychloroquine | Hair whitening (mainly in blonds/redheads) |
| Interferon (IFN) therapy | Whitening |
| Tyrosine kinase inhibitors (sunitinib, imatinib, dasatinib, cabozantinib) | Diffuse hypomelanosis |
| Triparanol | Hypopigmented hair |
| Minoxidil | Paradoxically darkens hair (converts vellus to terminal) |
F. Syndromes / Genetic Disorders
| Syndrome | Feature |
|---|---|
| Werner syndrome | Premature canities + cataracts, accelerated aging |
| Progeria (HGPS) | Premature greying in childhood |
| Rothmund-Thomson syndrome | Canities + photosensitivity, skeletal defects |
| Ataxia-telangiectasia | Premature greying + cerebellar ataxia, malignancy risk |
| Dyskeratosis congenita | Short telomere syndrome; premature greying + marrow failure |
| Waardenburg syndrome / Piebaldism | Poliosis (patchy white hair) |
| Myotonic dystrophy | Associated premature canities |
| Down syndrome, Fanconi syndrome | Associated diffuse hypomelanosis of scalp |
- Dermatology 2-Volume Set 5e, Tables 66.8 and 66.9; Andrews' Diseases of the Skin
Treatment
There is no universally effective treatment that reverses greying once melanocyte loss has occurred. However, several approaches address reversible causes and slow progression:
1. Treat Reversible Underlying Causes (Most Important)
- Iron supplementation: reverses canities segmentata sideropenica completely.
- Vitamin B12 replacement: IV or IM cyanocobalamin/methylcobalamin; repigmentation possible if melanocytes intact.
- Thyroid disease management: correct hypothyroidism or hyperthyroidism.
- Copper supplementation: for documented deficiency.
- Vitamin D correction
- Stop offending drugs (chloroquine, TKIs) if clinically feasible.
2. Antioxidant Therapy
Rationale: H₂O₂ accumulation and oxidative stress drive melanocyte damage. Several agents target this:
- Catalase-based topicals (PC-KUS/Suncat) - a pseudocatalase cream used with narrow-band UVB showed repigmentation in some vitiligo-associated greying cases.
- Oral antioxidants: vitamin E, vitamin C, selenium (supportive evidence only).
- Para-aminobenzoic acid (PABA) - historically used; some early evidence of partial repigmentation.
3. Phototherapy
- Narrow-band UVB (NB-UVB) - used for vitiligo-associated poliosis/greying; stimulates residual melanocytes.
- PUVA - less commonly used now due to side effects.
4. Topical Agents
- Minoxidil - indirectly darkens hair by converting vellus to terminal hairs.
- Topical prostaglandin analogues (latanoprost) - may pigment eyelashes.
5. Emerging / Investigational
- JAK inhibitors (e.g., ruxolitinib, baricitinib) - approved for alopecia areata; some repigmentation of greying hairs observed in alopecia areata patients as a secondary finding.
- Stem cell-based therapies targeting melanocyte stem cell niches are under research.
- Stress reduction and lifestyle modification (smoking cessation, balanced diet, antioxidant-rich foods) - may slow progression.
6. Cosmetic (Non-Medical)
- Hair dyes remain the most effective and widely used option for cosmetic management.
- Henna, semipermanent, and permanent dyes - patient preference guides selection.
Key Takeaway Summary
| Category | Key Points |
|---|---|
| Most common cause | Genetics/family history |
| Most reversible cause | Nutritional deficiency (iron, B12, copper) |
| Most important workup | CBC, ferritin, serum B12, TSH, vitamin D |
| Definitive treatment | None for true genetic canities |
| Partial reversal possible | In deficiency states, vitiligo (NB-UVB), alopecia areata (JAK inhibitors) |
A 2025 comprehensive review (Desai et al., Int J Dermatol 2025, PMID 39697103) confirmed genetics, oxidative stress, smoking, diet, and associated medical conditions as the main drivers, and highlighted that treatment remains limited - with the greatest benefit from identifying and correcting reversible causes.
Warner syndrome
Finding Sources
Searching PubMed
"Werner syndrome" AND (review OR management OR genetics)
Finding Sources
Reading Article
PMID: 39956107
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have thorough coverage from multiple authoritative textbooks. Here is the complete, detailed answer:
Werner Syndrome (Adult Progeria)
Overview
Werner syndrome (WS) is a rare autosomal recessive segmental progeroid syndrome - meaning it mimics many, but not all, features of normal aging. It manifests predominantly in the second and third decades of life and carries a significantly elevated cancer risk and shortened lifespan (~54 years on average). It was first described by Otto Werner in 1904.
Epidemiology
- Incidence: 1 in 1 million births worldwide.
- Much higher in Japan (1 in 3500 in some communities) due to consanguinity - most reported cases are Japanese.
- Affects both sexes equally.
- Over 800 patients reported worldwide.
- Dermatology 2-Volume Set 5e; Fitzpatrick's Dermatology
Genetics and Pathogenesis
- Caused by loss-of-function mutations in the WRN gene (RECQL2), located on chromosome 8p12.
- The WRN protein is a member of the RecQ helicase family and also has 3'→5' exonuclease activity.
- Functions of WRN protein:
- Optimizes DNA repair (especially base excision repair)
- Suppresses illegitimate recombination
- Maintains telomere integrity
- Regulates DNA replication fidelity
- Consequence of loss: Genomic instability accumulates, cells undergo premature senescence with reduced replicative capacity, leading to accelerated aging and malignant transformation.
- Over half of Japanese patients are homozygous for a specific splice-site mutation.
-
70 distinct pathogenic variants have been identified across the WRN locus.
- Atypical WS: A subset with identical phenotype carry heterozygous missense mutations in LMNA (lamin A/C gene) instead of WRN - these tend to have earlier onset and more severe manifestations, overlapping with Hutchinson-Gilford Progeria Syndrome (HGPS).
- Dermatology 2-Volume Set 5e; Paccosi et al., Cytogenet Genome Res, 2025 [PMID 39956107]
Clinical Features
Development is normal until early adolescence, after which the characteristic growth spurt fails to occur. Diagnosis is often delayed until the 4th or 5th decade.
Cutaneous (Skin/Hair) Features
- Premature greying and thinning of scalp hair (often beginning in childhood, characteristically in late adolescence/early 20s)
- Premature balding
- Scleroderma-like skin - tightening, atrophy, and induration of skin (most prominent on face, forearms, hands, legs, feet)
- Poikiloderma - mottled hyperpigmentation and hypopigmentation
- Loss of subcutaneous fat (lipodystrophy) and muscle wasting, especially in limbs
- Dystrophic, hypoplastic, or absent nails
- Plantar hyperkeratosis and thick keratoses over pressure points (fingers, toes, ankles, elbows, ears)
- Chronic, painful leg ulcers - particularly around malleoli, Achilles tendon, heels, toes; prone to infection due to ischaemia; resistant to treatment
- Dystrophic soft tissue calcifications
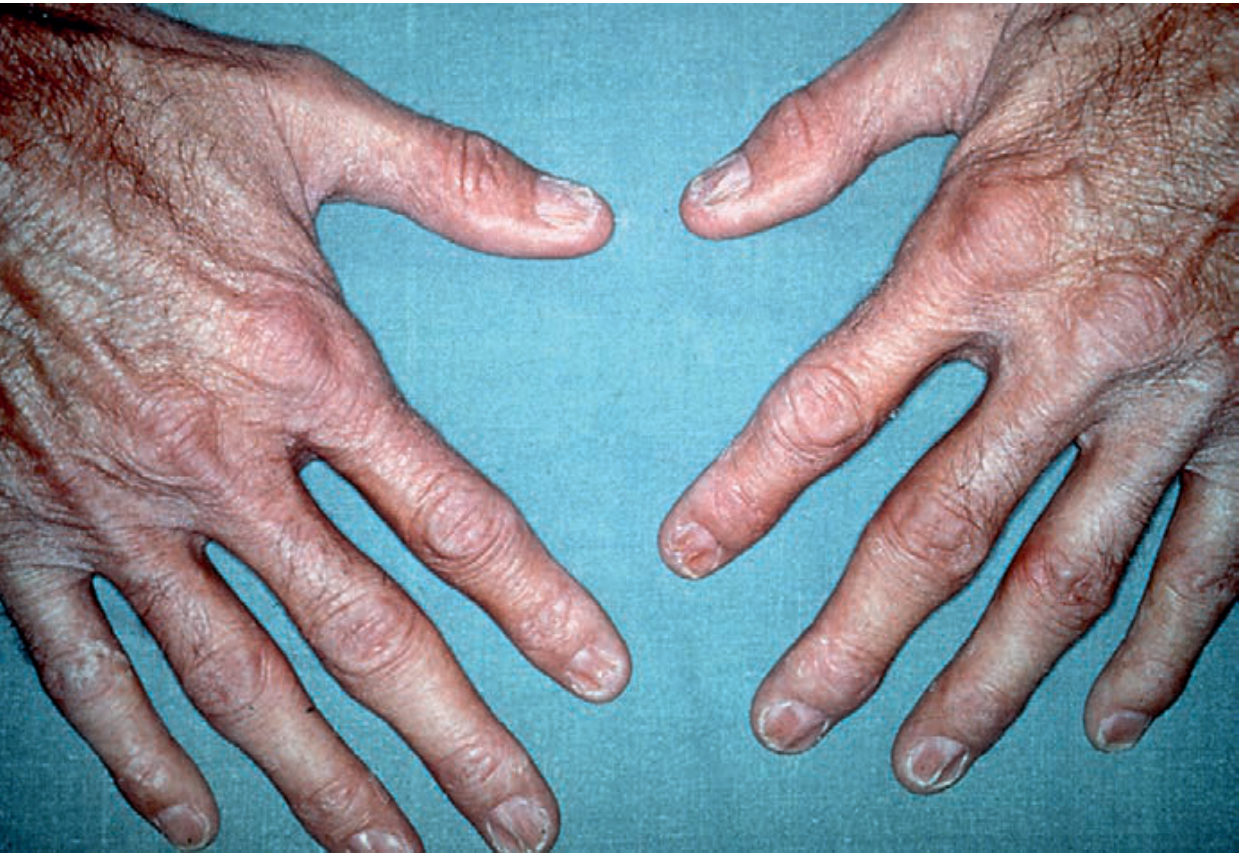
Werner syndrome - severe skin atrophy, prominent veins, dystrophic nails on the hands
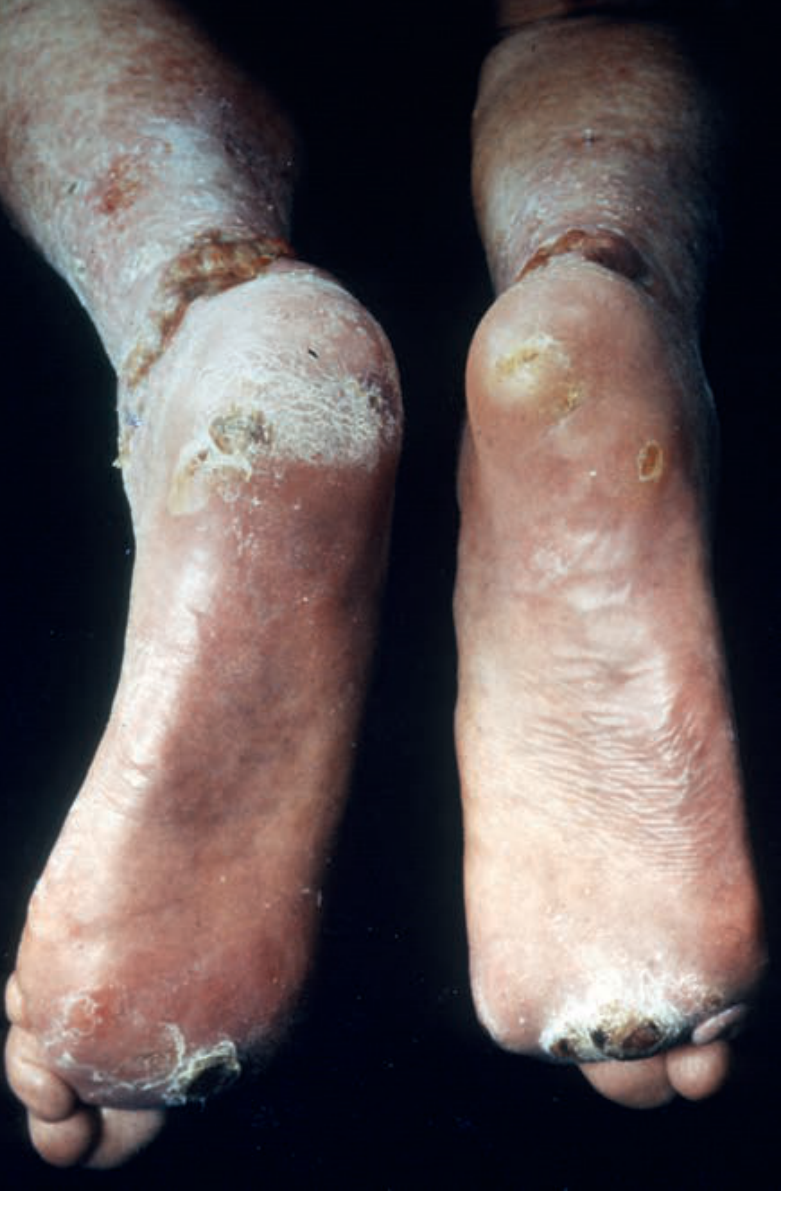
Werner syndrome - recalcitrant bilateral heel and leg ulcers, a major source of morbidity
Facial Features ("Bird-like Facies")
- Short stature (average height ~1.5 m / 5 ft), spindly limbs, central obesity ("trunk-stocky, extremities-thin" body habitus)
- Small hands and feet
- Thin, pinched face with prominent, proptotic eyes
- Beaked nose, taut lips, circumoral radial furrows
- Protuberant teeth, micrognathia
- High-pitched, hoarse/raspy voice (laryngeal atrophy/vocal cord thickening)
Ocular
- Bilateral posterior subcapsular cataracts - develop in the late 20s to 30s; ophthalmologists are often the first to suspect the diagnosis.
Metabolic / Internal Organ Features
| Feature | Notes |
|---|---|
| Type 2 diabetes mellitus | Very common; often insulin-resistant |
| Hyperlipidaemia | Elevated cholesterol, triglycerides |
| Generalised atherosclerosis | Premature, widespread; major cause of death |
| Hypertension | Less common than other features |
| Osteoporosis | Widespread; fracture risk |
| Osteosclerosis of distal extremities | Distinctive; NOT seen in normal aging |
| Hypogonadism | Both sexes; NOT a feature of normal aging |
| Aseptic necrosis of small hand bones | Common finding |
| Calcinosis circumscripta | Dystrophic calcifications, especially around joints |
Malignancy Risk
WS carries a dramatically elevated cancer risk with a pattern different from the general population:
- Normal aging shows a 10:1 epithelial:mesenchymal cancer ratio; WS shifts this to 1:1
- Especially increased in Japanese patients:
- 50-fold increase in melanoma (mostly acral-lentiginous or mucosal - not UV-related)
- Thyroid follicular carcinoma (at younger age than general population)
- Meningioma
- Soft tissue sarcomas, osteosarcoma, fibrosarcoma
- Hepatoma
- Leukemia
- Breast carcinoma
Prognosis
- Average lifespan: ~54 years (some patients die before age 50)
- Principal causes of death: cardiovascular/cerebrovascular disease and malignancy
- Heterozygous carriers may have elevated rates of malignancy and myocardial infarction compared to the general population.
Pathology / Histology
- Epidermis: hyperkeratosis, atrophy, focal basal layer hyperpigmentation
- Dermis: fibrosis and variable hyalinisation; collagen fibres parallel to epidermis (resembling scar/tendon)
- Appendages (hair follicles, sebaceous glands, eccrine glands): decreased in number, atrophic
- Subcutaneous fat: atrophic, often replaced by hyalinised connective tissue
- Blood vessels: changes typical of diabetic angiopathy
Diagnosis
Clinical diagnosis is made on recognition of the characteristic tetrad:
- Premature greying/canities
- Bird-like facies (beaked nose, micrognathia)
- Short stature
- Bilateral cataracts
Confirmatory tests:
- Genetic testing for WRN mutations (recommended when clinical features are inconclusive)
- Laboratory evaluation for diabetes, hyperlipidaemia, thyroid function
- Detailed diagnostic criteria available at the International Werner Syndrome Registry
Differential Diagnosis:
| Condition | Key Distinguishing Feature |
|---|---|
| Hutchinson-Gilford Progeria (HGPS) | Onset in infancy; no cataracts, no malignancy increase |
| Atypical WS (LMNA mutations) | Earlier onset, more severe; overlaps with HGPS |
| Cockayne syndrome | UV sensitivity, neurodegeneration, onset in childhood |
| Rothmund-Thomson syndrome | Photosensitivity, absent cataracts |
| MDPL syndrome (POLD1) | Hearing impairment, NO cataracts, NO malignancy |
| Ataxia-telangiectasia | Cerebellar ataxia, telangiectasia, immunodeficiency |
| Prolidase deficiency | Facial dysmorphism, telangiectasias, leg ulcers |
Treatment
There is no cure or specific disease-modifying therapy for Werner syndrome. Management is entirely supportive and symptomatic, aimed at complications.
1. Leg Ulcers (Major Source of Morbidity)
- Treat aggressively and early - debridement, wound care, infection control.
- Skin grafting may be required for resistant ulcers.
- Severe, recalcitrant disease may lead to lower limb amputation.
- Etidronate (bisphosphonate): case reports suggest it can ameliorate painful soft tissue calcifications.
2. Diabetes Mellitus
- Dietary management + appropriate medications:
- Thiazolidinediones (improve insulin sensitivity)
- Metformin
- DPP-4 inhibitors ("gliptins")
- Control reduces atherosclerosis complications.
3. Hyperlipidaemia
- Lipid-lowering agents (statins) to reduce cardiovascular risk.
4. Cataracts
- Surgical cataract extraction - usually successful; ophthalmologists often first to diagnose WS.
5. Osteoporosis
- Calcium, vitamin D, bisphosphonates as appropriate.
6. Cancer Surveillance
- Regular screening for thyroid cancer, melanoma (especially acral/mucosal), soft tissue sarcomas.
- Dermatological surveillance is important.
7. Genetic Counselling
- Offered to all affected families; prenatal diagnosis is possible when the familial mutation is known.
8. Experimental / Emerging Therapies (Under Investigation)
- Vitamin C supplementation: reversed age-related metabolic abnormalities and increased lifespan in a mouse WS model - may be beneficial in humans.
- mTOR inhibitors (rapamycin analogues): under investigation for reducing cellular senescence.
- CRISPR/Cas9-mediated gene correction: experimental; potential future therapeutic direction.
- Dermatology 2-Volume Set 5e; Andrews' Diseases of the Skin; Fitzpatrick's Dermatology; Paccosi et al., Cytogenet Genome Res 2025 [PMID 39956107]
Quick Summary
| Feature | Detail |
|---|---|
| Gene | WRN (RECQL2) - RecQ helicase + exonuclease |
| Inheritance | Autosomal recessive |
| Onset | 2nd decade (growth failure); full picture 3rd-4th decade |
| Classic tetrad | Premature greying + bird facies + short stature + cataracts |
| Unique features | Hypogonadism, laryngeal atrophy, osteosclerosis of extremities |
| Major complications | Atherosclerosis, T2DM, malignancy (especially sarcomas, melanoma) |
| Lifespan | ~54 years |
| Cause of death | Cardiovascular disease or malignancy |
| Treatment | Symptomatic only; no cure |
This is a shared conversation. Sign in to Orris to start your own chat.