Conjugated hyperbillirumenia
Reading File
Finding Sources
Finding Sources
Searching PubMed
"conjugated hyperbilirubinemia" causes diagnosis
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Conjugated Hyperbilirubinemia
Definition
Conjugated (direct) hyperbilirubinemia is defined as a serum conjugated bilirubin >2 mg/dL, or >20% of total bilirubin. Unlike unconjugated hyperbilirubinemia, it is never physiologic and always indicates hepatocellular disease, biliary obstruction, or a defect in canalicular excretion. Because conjugated bilirubin is water-soluble, it appears in the urine (bilirubinuria/choluria) - this is a bedside clue.
Bilirubin Metabolism - Quick Review
| Step | Location | Event |
|---|---|---|
| Heme breakdown | Reticuloendothelial system | Heme → unconjugated (indirect) bilirubin + albumin complex |
| Uptake | Hepatocyte sinusoidal membrane (OATP1B1/B3) | Bilirubin-albumin taken up by hepatocytes |
| Conjugation | Hepatocyte ER | UGT1A1 converts to bilirubin diglucuronide (water-soluble) |
| Excretion | Bile canaliculus (MRP2/ABCC2) | Conjugated bilirubin excreted into bile |
| Intestinal fate | Gut bacteria | Urobilinogen → stercobilin (stool color) + urobilinogen reabsorbed |
Causes - Classified by Mechanism
1. Hereditary / Isolated (Normal other LFTs)
These are benign autosomal recessive disorders with normal hepatic function:
Dubin-Johnson Syndrome (DJS)
- Defect: Mutation in ABCC2 gene encoding the canalicular export pump MRP2
- Consequence: Conjugated bilirubin cannot enter the bile canaliculus; it regurgitates back into blood
- Serum bilirubin: typically 2-5 mg/dL (can reach 20-25 mg/dL during illness, OCP use, or pregnancy)
- Histology: Black coarsely granular pigment (epinephrine metabolites) in centrilobular hepatocyte lysosomes - the liver may appear grossly black
- Urine coproporphyrin: Total normal, but coproporphyrin I = 80% (normally coproporphyrin III predominates)
- All other LFTs: Normal
- Prognosis: Benign
Rotor Syndrome
- Defect: Mutations in SLCO1B1 and SLCO1B3 (encoding OATP1B1 and OATP1B3) - impaired hepatic re-uptake of conjugated bilirubin secreted by MRP3 into plasma
- No black pigment on biopsy
- Urine coproporphyrin: Total elevated 2-5x; both I and III elevated
- Similarly benign
| Feature | Dubin-Johnson | Rotor |
|---|---|---|
| Defective protein | MRP2 (canalicular export) | OATP1B1 + OATP1B3 (sinusoidal uptake) |
| Liver pigment | Dark black granules | Absent |
| Total urine coproporphyrin | Normal | Elevated 2-5x |
| Coproporphyrin I fraction | ~80% | ~65% |
| Serum bilirubin | 2-5 mg/dL | 3-7 mg/dL |
(Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine 22E)
2. Hepatocellular Disease (Mixed / Predominantly Conjugated)
In acquired liver disease, both conjugated and unconjugated bilirubin typically rise together. The major categories:
- Viral hepatitis (HAV, HBV, HCV, HDV, HEV) - HAV/HEV are enteral and self-limited; HBV/HCV/HDV are parenteral and may become chronic
- Drug-induced liver injury (DILI) - acetaminophen overdose is most common toxic cause; idiosyncratic reactions with many other drugs
- Alcoholic hepatitis / cirrhosis
- Autoimmune hepatitis
- Ischemic hepatitis (shock liver)
Key lab pattern: Elevated ALT/AST (hepatocellular injury pattern), conjugated + unconjugated bilirubin both elevated, elevated GGT and ALP in cholestatic component.
3. Cholestatic Disorders
Intrahepatic cholestasis:
- Primary biliary cholangitis (PBC) - autoimmune destruction of small bile ducts; anti-mitochondrial antibody positive
- Primary sclerosing cholangitis (PSC) - fibro-obliterative disease of large ducts; associated with IBD
- Drug-induced cholestasis (e.g., estrogens, cyclosporine, chlorpromazine)
- Intrahepatic cholestasis of pregnancy
- Progressive familial intrahepatic cholestasis (PFIC) - mutations in bile transport genes (FIC1, BSEP, MDR3)
- Infiltrative disorders (sarcoidosis, lymphoma, amyloid, metastases)
- Sepsis-associated cholestasis
Extrahepatic (obstructive) cholestasis:
- Choledocholithiasis (gallstone in common bile duct) - most common cause
- Cancer of the head of pancreas - classic painless obstructive jaundice
- Cholangiocarcinoma
- Periampullary carcinoma
- Biliary strictures (post-surgical, post-inflammatory)
Key lab pattern: ALP and GGT disproportionately elevated vs. aminotransferases; pale stools, dark urine, pruritus.
(Harper's Illustrated Biochemistry 32nd Ed; Robbins & Cotran Pathologic Basis of Disease)
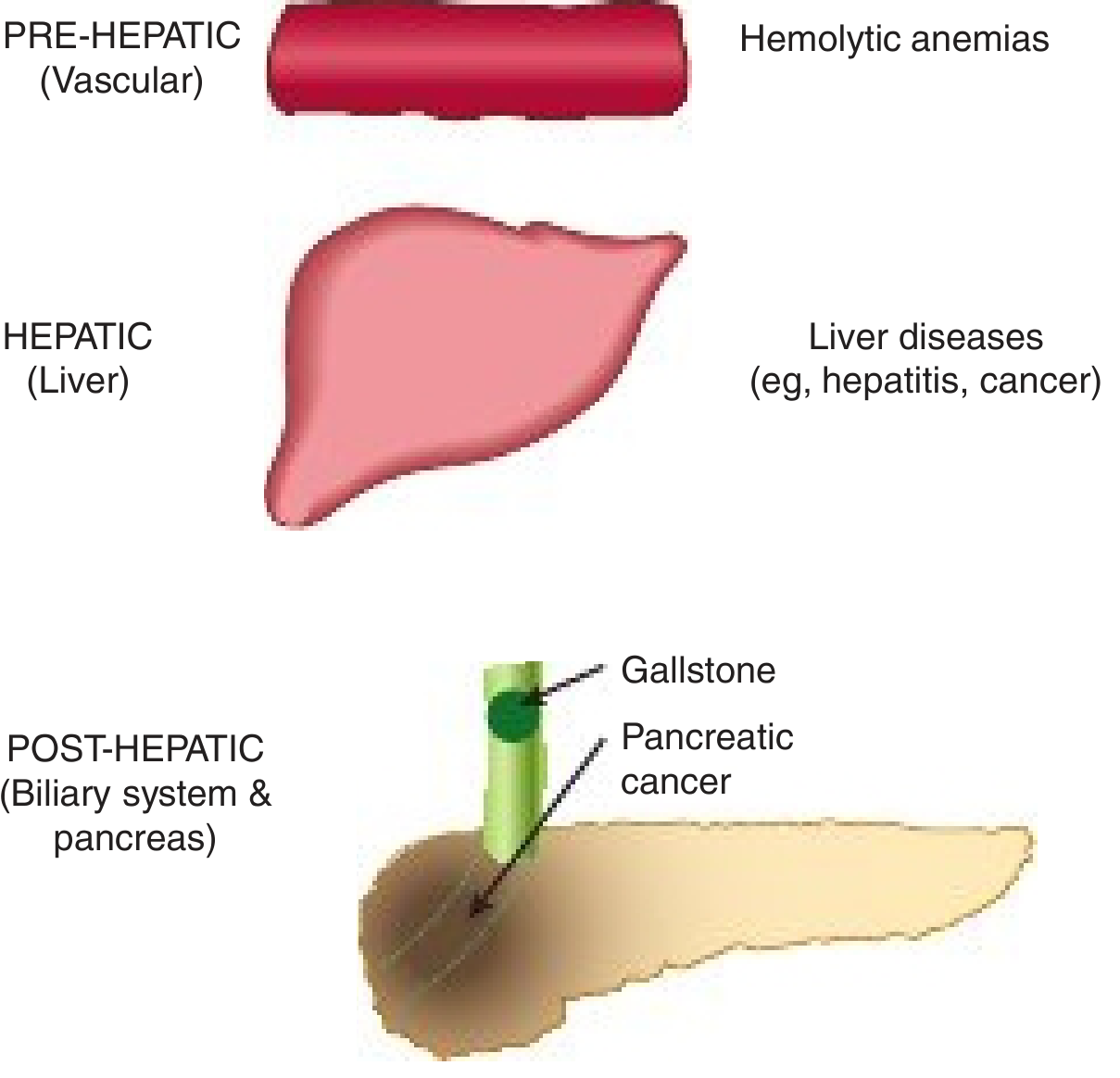
4. Neonatal / Pediatric Causes
Neonatal cholestasis - defined as prolonged conjugated hyperbilirubinemia in a neonate; affects ~1 in 2,500 live births. Always pathologic (contrast with physiologic neonatal jaundice which is unconjugated). Major causes:
- Biliary atresia - progressive fibro-obliteration of bile ducts; requires Kasai portoenterostomy (surgical); leading cause of pediatric liver transplant
- Neonatal hepatitis - a syndromic diagnosis encompassing toxic, metabolic, genetic, and infectious causes; >85% have identifiable etiology
- Infectious: CMV, rubella, syphilis, toxoplasmosis, HSV
- Metabolic: alpha-1-antitrypsin deficiency, galactosemia, tyrosinemia, Niemann-Pick
- Idiopathic (~10-15%)
Differentiating biliary atresia from neonatal hepatitis is critical since BA requires surgery but other causes may be worsened by it. Liver biopsy may be needed in ~10% of cases. (Robbins Basic Pathology)
Clinical Features of Conjugated Hyperbilirubinemia
| Feature | Mechanism |
|---|---|
| Jaundice (yellow skin/sclera) | Tissue deposition of bilirubin |
| Dark urine (bilirubinuria) | Water-soluble conjugated bilirubin filtered by kidney |
| Pale/acholic stools | Reduced bilirubin reaching gut |
| Pruritus | Accumulation of bile salts in skin |
| Skin xanthomas | Cholesterol accumulation |
| Fat malabsorption | Reduced bile in gut → deficiency of fat-soluble vitamins (A, D, E, K) |
| Elevated ALP + GGT | Enzymes on canalicular membranes of hepatocytes and bile duct epithelia |
(Robbins & Cotran Pathologic Basis of Disease, p. 793)
Key Lab Pattern Comparison
| Type | Conjugated bili | Unconjugated bili | ALP/GGT | ALT/AST | Urine bilirubin | Urine urobilinogen |
|---|---|---|---|---|---|---|
| Hemolytic | Normal | ↑↑ | Normal | Normal | Absent | ↑↑ |
| Hepatocellular (e.g., hepatitis) | ↑ | ↑ | ↑ | ↑↑ | Present | ↓ (if microobstruction) |
| Obstructive (cholestasis) | ↑↑ | Normal/slight ↑ | ↑↑ | Normal/slight ↑ | Present | Absent |
| DJS/Rotor | ↑ | Normal | Normal | Normal | Present | Normal |
(Harper's Illustrated Biochemistry 32nd Ed, Table 31-4)
Pathology of Cholestasis
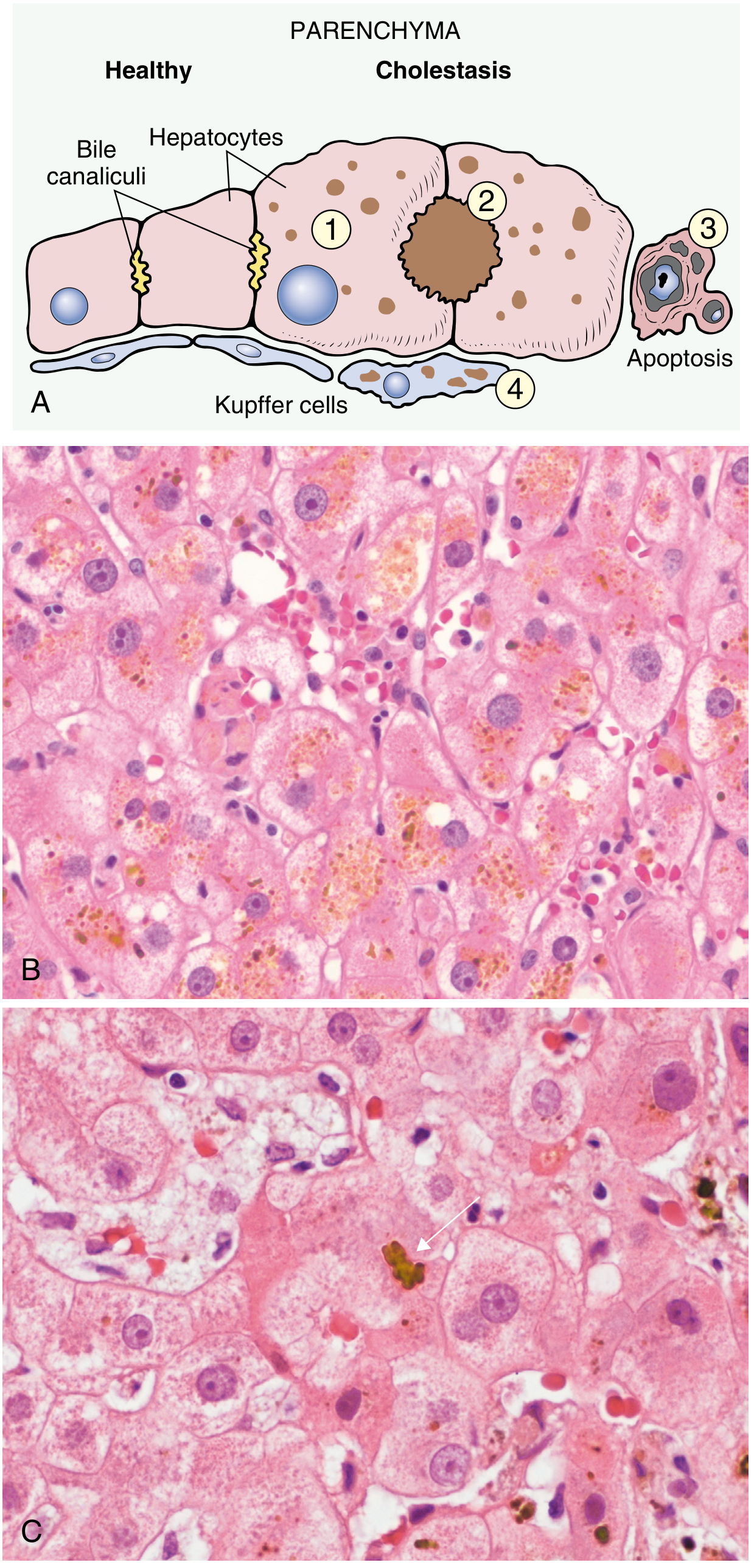
On histology, cholestasis shows:
- Enlarged hepatocytes with dilated canalicular spaces
- Bile plugs in expanded bile canaliculi
- Kupffer cells loaded with regurgitated bile pigment
- Hepatocyte apoptosis
- In chronic obstruction: biliary cirrhosis
Important Clinical Points
- Bilirubinuria always = conjugated hyperbilirubinemia (unconjugated bilirubin is albumin-bound and not filtered by kidneys)
- In prolonged conjugated hyperbilirubinemia, bilirubin can become covalently bound to albumin (delta bilirubin). This has a long half-life (~18 days like albumin) and persists after resolution of the underlying disease - patients may remain mildly jaundiced even as cholestasis resolves
- DJS and Rotor syndrome are the only causes of isolated conjugated hyperbilirubinemia with completely normal all other liver function tests
- Distinguishing intrahepatic from extrahepatic cholestasis by bilirubin fractions alone is unreliable - imaging (ultrasound, MRCP) is necessary
Sources: Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine 22E (2025); Robbins & Cotran Pathologic Basis of Disease; Robbins Basic Pathology; Harper's Illustrated Biochemistry 32nd Ed; Sleisenger & Fordtran's Gastrointestinal and Liver Disease
Hyperurecimia
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive material. Let me compile the full answer.
Hyperuricemia
Definition
Hyperuricemia is defined as a serum urate concentration >6.8 mg/dL (408 µmol/L) at physiologic pH (7.40) and normal body temperature - the saturation point of monosodium urate (MSU) in plasma. Above this threshold, urate can precipitate as MSU crystals. Hyperuricemia is a necessary (but not sufficient) prerequisite for gout; only ~20% of hyperuricemic individuals ever develop gout. (Goldman-Cecil Medicine)
Biochemical Basis - Purine Metabolism
Uric acid is the end product of purine catabolism in humans. The pathway:
AMP / GMP
↓ (5'-nucleotidase)
Adenosine / Guanosine / Inosine
↓ (purine nucleoside phosphorylase)
Hypoxanthine / Guanine
↓ (xanthine oxidase)
Xanthine
↓ (xanthine oxidase)
Uric acid
Key facts:
- Xanthine oxidase (in liver + intestine) catalyzes the final two steps - the drug target for allopurinol and febuxostat
- Most mammals possess uricase which converts uric acid → allantoin (highly soluble). Humans and other great apes lost uricase ~18 million years ago, making urate accumulation possible
- ~2/3 of daily urate turnover is excreted by kidneys; ~1/3 by gut (intestinal uricolysis)
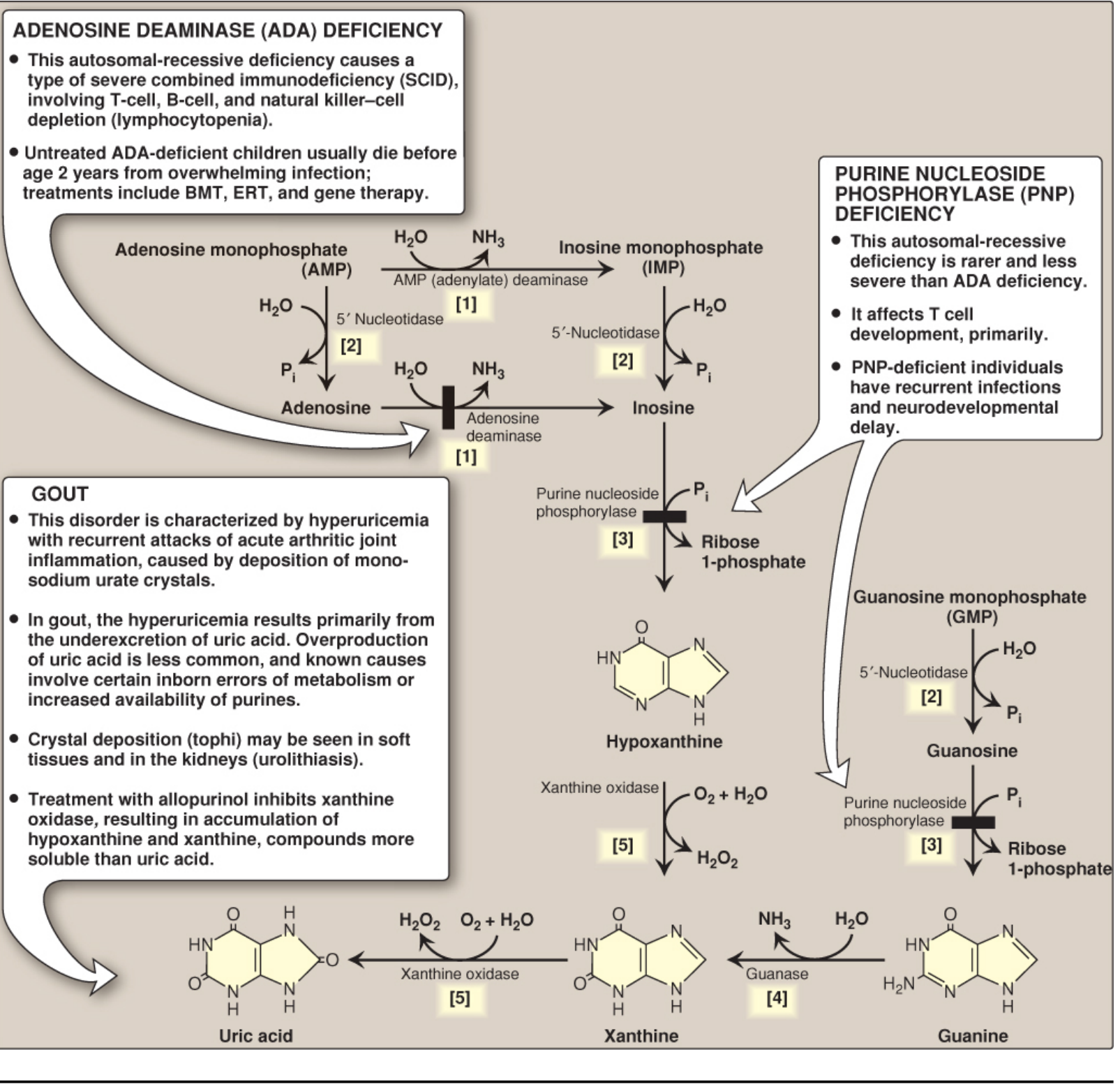
(Lippincott Illustrated Reviews Biochemistry, 8th Ed)
Renal Handling of Urate
Uric acid is small, unbound, and completely filtered at the glomerulus. Under normal conditions, 90% of filtered urate is reabsorbed in the proximal convoluted tubule via the transportasome - a collection of organic acid transporters, the most important being URAT1.
URAT1 performs a urate-monocarboxylate exchange. It can be driven to reabsorb more urate when tubular lactate, pyruvate, ketoacids (acetoacetate, β-hydroxybutyrate) are elevated - explaining hyperuricemia in starvation, alcoholism, and diabetic ketoacidosis.
Causes of Hyperuricemia
Mechanism-Based Classification
| Mechanism | Fraction of Cases |
|---|---|
| Renal underexcretion (primary/secondary) | >90% |
| Overproduction of uric acid | <10% |
| Combined underexcretion + overproduction | Small subset |
A. Underexcretion (Renal)
Gouty patients excrete only 70% as much uric acid as normal individuals at the same serum urate level; they require a serum urate 1.7 mg/dL higher to achieve equivalent excretion.
Non-genetic (secondary) causes - impaired excretion:
| Category | Examples |
|---|---|
| Reduced GFR | CKD of any cause |
| Hypertension | Direct tubular effect + decreased renal blood flow |
| Obesity / Metabolic syndrome | Insulin resistance reduces renal urate excretion |
| Volume depletion | Increased tubular reabsorption of all solutes including urate |
| Acidosis | Lactate, ketoacids, and other organic acids compete with URAT1, driving urate reabsorption |
| Drugs | Thiazide diuretics (most common drug cause), loop diuretics, low-dose aspirin (<2 g/day), cyclosporine, tacrolimus, pyrazinamide, ethambutol, nicotinic acid, levodopa |
| Lead nephropathy (saturnine gout) | Tubular damage impairing urate secretion |
| Hypothyroidism | Reduced GFR and tubular effects |
| Primary hyperparathyroidism | Reduced renal urate clearance |
| Organ transplantation (CsA/tacrolimus use) |
Genetic causes - impaired excretion:
- Mutations in URAT1 (SLC22A12) - gain-of-function → increased reabsorption → familial gout
- GLUT-9 (SLC2A9) polymorphisms - the single most significant genetic determinant of serum urate
- ABCG2 mutations - intestinal/renal urate exporter; loss-of-function → up to 10% of gout in White Europeans
- Familial juvenile hyperuricemic nephropathy (UMOD mutations) - early-onset gout + progressive CKD
B. Overproduction
Non-genetic (secondary) causes - overproduction:
| Condition | Mechanism |
|---|---|
| Myeloproliferative / lymphoproliferative disorders | Accelerated cell turnover → increased purine load |
| Tumor lysis syndrome | Massive nucleic acid release |
| Chronic hemolytic anemia | Increased red cell turnover |
| Polycythemia vera | Increased cell turnover |
| Psoriasis | Increased epidermal cell turnover |
| Alcohol use disorder | Accelerated purine nucleotide degradation; also impairs excretion via lactate |
| High-fructose intake | Fructose-1-phosphate traps phosphate → AMP accumulation → urate production |
| Vigorous exercise | Muscle ATP breakdown → AMP → urate |
Genetic/primary causes - overproduction:
- Lesch-Nyhan Syndrome - X-linked recessive; complete deficiency of HPRT (hypoxanthine-guanine phosphoribosyl transferase); no purine salvage → PRPP accumulates → de novo synthesis floods → marked hyperuricemia + self-mutilation + choreoathetosis + intellectual disability
- Kelley-Seegmiller Syndrome - partial HPRT deficiency → milder disease, gout without neurologic features
- PRPP Synthetase Overactivity - X-linked; superactive enzyme with increased Vmax, decreased Km for ribose-5-phosphate, or decreased feedback sensitivity to purine nucleotides → excess PRPP → excess de novo synthesis
- Von Gierke Disease (GSD type Ia) - G6Pase deficiency; shunts glucose-6-phosphate into pentose phosphate pathway → excess PRPP + lactic acidosis (blocks renal excretion)
- Hereditary fructose intolerance - fructose-1-phosphate accumulation traps phosphate → AMP degradation
Monosodium Urate Crystal Formation and Gout
Once serum urate exceeds 6.8 mg/dL, MSU crystals can form, but this is NOT inevitable. Factors that promote crystal formation:
- Supersaturated urate concentration (sustained)
- Low pH and low temperature (favors peripheral joints - first MTP, Achilles)
- Seed nuclei (cartilage debris, collagen, chondroitin)
- Immunoglobulins IgG and IgM (serve as crystal nucleation platforms)
- Intra-articular dehydration
- Osteoarthritic cartilage degradation products (lower urate solubility)
Gout flare mechanism:
- MSU crystals in synovial space coated by IgG → phagocytosis by neutrophils/macrophages
- Activation of NLRP3 inflammasome → caspase-1 activation → IL-1β processing and secretion
- IL-1β → neutrophil recruitment, endothelial activation (E-selectin, ICAM-1), cytokine/chemokine cascade
- Apolipoprotein B/E coating of crystals inhibits this response - explains why crystals can exist without inflammation (intercritical gout)
Clinical Spectrum
1. Asymptomatic Hyperuricemia
- No symptoms; not a disease in itself
- Risk of clinical gout rises steeply with urate level: incidence 4.9%/year at SU >9 mg/dL vs. 0.5%/year at 7-8.9 mg/dL
- 50% of those with SU >10 mg/dL eventually develop clinical gout
- Comorbidities: metabolic syndrome, obesity, dyslipidemia, hypertension, DM, CKD, congestive heart failure
2. Acute Gout Flare
- Explosive onset of severe joint pain, reaching maximum intensity in 12-24 hours
- Classic: first metatarsophalangeal joint (podagra), ankle, midfoot, knee
- Erythema, warmth, exquisite tenderness ("worst pain ever")
- Spontaneously resolves in days-weeks if untreated
- Triggered by: rapid change in urate level (initiation of ULT, dehydration, illness, alcohol, surgery)
3. Intercritical Gout
- Asymptomatic periods between flares
- MSU crystals may still be detectable in joints on DECT/ultrasound even asymptomatically
4. Chronic Tophaceous Gout
- Nodular MSU crystal deposits (tophi) in soft tissues: pinnae of ears, olecranon bursa, Achilles tendon, finger pads
- Chronic joint destruction and deformity
- Can also develop in axial skeleton, heart valves, kidneys
5. Uric Acid Nephrolithiasis
- MSU / uric acid crystals in collecting system
- Associated with hyperuricemia, low urine pH, concentrated urine
- Produces radiolucent stones on plain X-ray (uric acid stones are not radio-opaque)
6. Urate Nephropathy
- Chronic: MSU crystal deposition in renal interstitium → tubular damage + CKD
- Acute: massive uric acid precipitation in tubules (tumor lysis syndrome) → oliguric AKI
Diagnosis
| Investigation | Finding |
|---|---|
| Serum urate | >6.8 mg/dL (may be normal or even low during acute flare) |
| 24-hr urinary uric acid | >1000 mg/day on normal diet = overproducer; <800 mg/day = underexcretion |
| Synovial fluid microscopy (gold standard) | Needle-shaped, negatively birefringent MSU crystals under polarized light |
| Serum urate during flare | May be falsely normal (flare lowers serum urate transiently) |
| DECT / Ultrasound | Detect tophi and synovial MSU deposits before clinically apparent |
Management
Non-pharmacologic
- Dietary: reduce red meat, organ meats, shellfish, high-fructose corn syrup; limit alcohol (especially beer/spirits)
- Increase low-fat dairy, cherry consumption, hydration
- Weight loss, manage metabolic syndrome
- Avoid precipitating drugs where possible (thiazides → switch to losartan which is uricosuric; aspirin → avoid low-dose in gouty patients)
Acute Flare Treatment (Targets: pain and inflammation)
| Drug | Mechanism | Notes |
|---|---|---|
| NSAIDs (indomethacin, naproxen) | COX inhibition | First-line; avoid in CKD, GI disease |
| Colchicine | Inhibits microtubule polymerization → inhibits neutrophil migration and NLRP3 inflammasome | Most effective when started within 24h of onset; low-dose as effective as high-dose with fewer side effects |
| Glucocorticoids (oral/intra-articular) | Anti-inflammatory | When NSAIDs/colchicine contraindicated |
| IL-1 inhibitors (anakinra, canakinumab) | Block IL-1β | Refractory cases |
Urate-Lowering Therapy (ULT) - Targets: serum urate <6 mg/dL (or <5 mg/dL in tophaceous gout)
| Drug | Class | Mechanism |
|---|---|---|
| Allopurinol | Xanthine oxidase inhibitor | Inhibits conversion of hypoxanthine/xanthine → uric acid; first-line ULT; start low, titrate |
| Febuxostat | Selective xanthine oxidase inhibitor | Alternative; non-purine structure; metabolized by liver (useful in mild-mod CKD) |
| Probenecid | Uricosuric agent | Blocks URAT1 → reduces tubular reabsorption; avoid in urolithiasis; ineffective in CKD |
| Losartan | Angiotensin receptor blocker | Mild uricosuric effect; useful in hypertensive gouty patients |
| Lesinurad | URAT1 + OAT4 inhibitor | Combination with XOI for refractory cases |
| Pegloticase | Recombinant uricase (PEGylated) | Converts urate → allantoin; for refractory tophaceous gout; IV infusion; risk of infusion reactions |
Important: Do NOT start ULT during an acute flare (can prolong or trigger new flares by rapidly changing serum urate). Co-prescribe colchicine or NSAIDs as prophylaxis for the first 3-6 months of ULT initiation.
Key Associations and Comorbidities
- Hypertension (bidirectional - urate promotes endothelial dysfunction and renin-angiotensin activation)
- Metabolic syndrome (insulin resistance reduces renal urate clearance)
- CKD (reduced GFR → hyperuricemia; hyperuricemia → tubular damage → worsens CKD)
- Cardiovascular disease (hyperuricemia is an independent risk factor; controversial if causative)
- Lead exposure (saturnine gout) - occupational or environmental
Sources: Goldman-Cecil Medicine 2-Vol Set; Rheumatology 2-Vol Set (Elsevier 2022); Firestein & Kelley's Textbook of Rheumatology; Lippincott Illustrated Reviews Biochemistry 8th Ed; Brenner & Rector's The Kidney; Tietz Textbook of Laboratory Medicine 7th Ed
This is a shared conversation. Sign in to Orris to start your own chat.