Pathophysiology of amyloidosis
Reading File
Finding Sources
Searching PubMed
amyloidosis pathophysiology mechanisms
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I now have all the information needed for a comprehensive answer. Let me compile the full response.
Pathophysiology of Amyloidosis
What is Amyloidosis?
Amyloidosis is not a single disease but a group of disorders defined by the extracellular deposition of insoluble protein fibrils that disrupt normal tissue architecture and function. The fibrils share a common structural core - a beta-pleated sheet configuration - regardless of which precursor protein generated them.
Core Molecular Mechanism: Protein Misfolding and Fibril Formation
The "amyloid hypothesis" proposes the following sequence:
- Precursor protein overproduction or reduced clearance - Systemic or local factors increase the concentration of an amyloidogenic precursor protein (e.g., immunoglobulin light chains, serum amyloid A, transthyretin).
- Misfolding/unfolding - The normally soluble precursor undergoes reversible unfolding, driven by inherited mutations, proteolytic clipping, aging, or microenvironmental factors (pH, metal ions, glycosaminoglycans).
- Oligomerization - Misfolded monomers self-assemble into small oligomers. These oligomers are now considered the most cytotoxic species - they interact directly with cell membranes, induce reactive oxygen species (ROS) formation, and trigger stress-signaling cascades.
- Fibril elongation - Oligomers stack into higher-order polymers and ultimately into mature, insoluble fibrils 7-10 nm in diameter with a characteristic cross-beta structure.
- Tissue deposition - Fibrils deposit in the extracellular matrix, co-localizing with serum amyloid P component (SAP), apolipoproteins E and A-IV, glycosaminoglycans (heparan sulfate), and metal ions. SAP protects fibrils from proteolytic degradation, stabilizing deposits.
- Organ dysfunction - Mechanical disruption of parenchymal architecture, direct oligomer toxicity, and activation of the unfolded protein response (UPR) all contribute to cell loss and organ failure.
"Accumulating evidence suggests that the oligomeric intermediates may constitute the most toxic species. Oligomers are more capable than fibrils of interacting with cells and inducing formation of reactive oxygen species and stress signaling." - Harrison's Principles of Internal Medicine, 22E
Classification by Precursor Protein
The standard nomenclature is AX, where A = amyloidosis and X = abbreviated precursor protein. From Harrison's 22E:
| Type | Precursor Protein | Clinical Setting | Primary Organs |
|---|---|---|---|
| AL | Immunoglobulin light chain (lambda > kappa) | Plasma cell dyscrasia (myeloma, lymphoma) | Any - heart, kidney, liver, nerves |
| AA | Serum amyloid A (SAA) | Chronic inflammation/infection | Kidney, liver, heart |
| ATTR (mutant) | Transthyretin (TTR) - mutated | Hereditary familial amyloidosis | Peripheral/autonomic nerves, heart |
| ATTR (wild-type) | TTR - normal | Age-related, usually males >65 | Heart (cardiomyopathy) |
| Aβ2M | Beta-2 microglobulin | Long-term hemodialysis | Synovial tissue, bone, joints |
| Aβ | Amyloid beta protein (APP cleavage) | Alzheimer's disease | CNS |
| AIAPP | Islet amyloid polypeptide (amylin) | Type 2 diabetes | Pancreatic islets |
Pathophysiology of Major Types
1. AL Amyloidosis (Most common systemic type in North America)
- A clonal expansion of bone marrow plasma cells secretes monoclonal immunoglobulin light chains (predominantly lambda subtype).
- The primary sequence of the light chain determines whether it misfolds (causing AL amyloidosis) or folds correctly (leading to multiple myeloma instead).
- Full-length 23-kDa light chains or their fragments form fibrils that deposit in the interstitium of virtually any organ outside the CNS.
- Accessory co-deposited molecules (SAP, ApoE, glycosaminoglycans) stabilize the fibrils.
- Cardiac direct proteotoxicity from soluble light chain oligomers contributes significantly to the rapid deterioration seen in AL cardiac amyloidosis, independent of fibril load.
2. AA Amyloidosis (Reactive/Secondary)
- Driven by sustained elevation of serum amyloid A (SAA), an acute-phase protein synthesized primarily in the liver under the influence of IL-1, TNF-alpha, and IL-6 (via NF-kB).
- Normal SAA levels: <1 mg/L; during acute inflammation: up to 1800 mg/L.
- Monocyte/macrophage-lineage cells cleave SAA via metalloproteinase activity, removing the stabilizing C-terminus. The exposed N-terminus is highly amyloidogenic and initiates aggregation.
- Deposited subunit fibrils are 5-8 kDa (truncated at the carboxy-terminal end).
- Genetic susceptibility matters: SAA1.1 homozygosity (UK) and SAA1.3 homozygosity (Japan) confer higher risk. A SNP at position -13 of the SAA1 gene also increases risk.
- Primary target organ: kidney (proteinuria, nephrotic syndrome, progressive renal failure).
3. ATTR Amyloidosis
- Transthyretin (TTR) is a 127-amino acid homo-tetrameric transport protein for thyroxine and retinol-binding protein, made primarily in the liver.
- Amyloidogenesis begins with tetramer dissociation into monomers, which misfold and aggregate. More than 100 TTR mutations destabilize the tetramer and accelerate misfolding.
- The V30M mutation is the most common hereditary variant (ATTRv), causing peripheral and autonomic neuropathy predominantly.
- Wild-type ATTR (ATTRwt) - formerly "senile systemic amyloidosis" - affects predominantly elderly men (>65 years) with cardiac infiltrative cardiomyopathy and carpal tunnel syndrome.
- TTR stabilizers (e.g., tafamidis) work by binding the T4-binding site, preventing tetramer dissociation and misfolding.
4. Aβ2M Amyloidosis (Dialysis-related)
- Beta-2-microglobulin (11.8 kDa), the invariant chain of MHC class I, is normally excreted by the kidney.
- In end-stage renal disease, it accumulates in the plasma (some dialysis membranes cannot clear it).
- Deposits preferentially in synovial tissue, periarticular structures, and bone - causing carpal tunnel syndrome, destructive arthropathy, and cystic bone lesions.
- Incidence is declining with high-flux dialysis membranes.
Mechanisms of Tissue Injury
Three converging mechanisms cause organ dysfunction:
-
Mechanical disruption - Bulk fibril deposits in the extracellular matrix physically displace and compress parenchymal cells. In the glomerulus, this leads to proteinuria and progressive nephron loss. In the heart, it causes ventricular wall stiffening (restrictive/infiltrative cardiomyopathy) with diastolic dysfunction.
-
Direct oligomer toxicity (proteotoxicity) - Pre-fibrillar oligomers are membrane-active, generating ROS, activating stress kinases (JNK, p38 MAPK), and triggering apoptosis. This mechanism is particularly important in AL cardiac amyloidosis.
-
Unfolded Protein Response (UPR) activation - ER stress from accumulating misfolded proteins triggers UPR, which if unresolved, activates apoptotic cascades (CHOP, caspase-12 pathway).
Additional mechanisms in specific contexts:
- Vascular involvement: Amyloid deposition in blood vessel walls impairs vasoconstriction, leading to orthostatic hypotension and hemorrhage.
- Factor X deficiency: AL amyloid deposits adsorb factor X, causing an acquired coagulopathy.
- Autonomic neuropathy: TTR fibrils damage small unmyelinated autonomic nerve fibers, causing gastroparesis, orthostatic hypotension, and bowel dysmotility.
Histopathological Hallmarks
- Congo red stain: Amyloid deposits stain brick-red; under polarized light, they display pathognomonic apple-green birefringence.
- Electron microscopy: Rigid, non-branching fibrils ~7-10 nm in diameter.
- Other stains: Thioflavin T/S (fluorescence), crystal violet (metachromasia).
- The regular beta-sheet structure is what generates the distinctive optical properties.
Diagnostic Algorithm
The diagnostic algorithm from Harrison's 22E (shown below) begins with noninvasive biopsy (abdominal fat, Congo red stain - positive in >80% of systemic cases), followed by mass spectrometry or immunohistochemistry to type the amyloid protein:
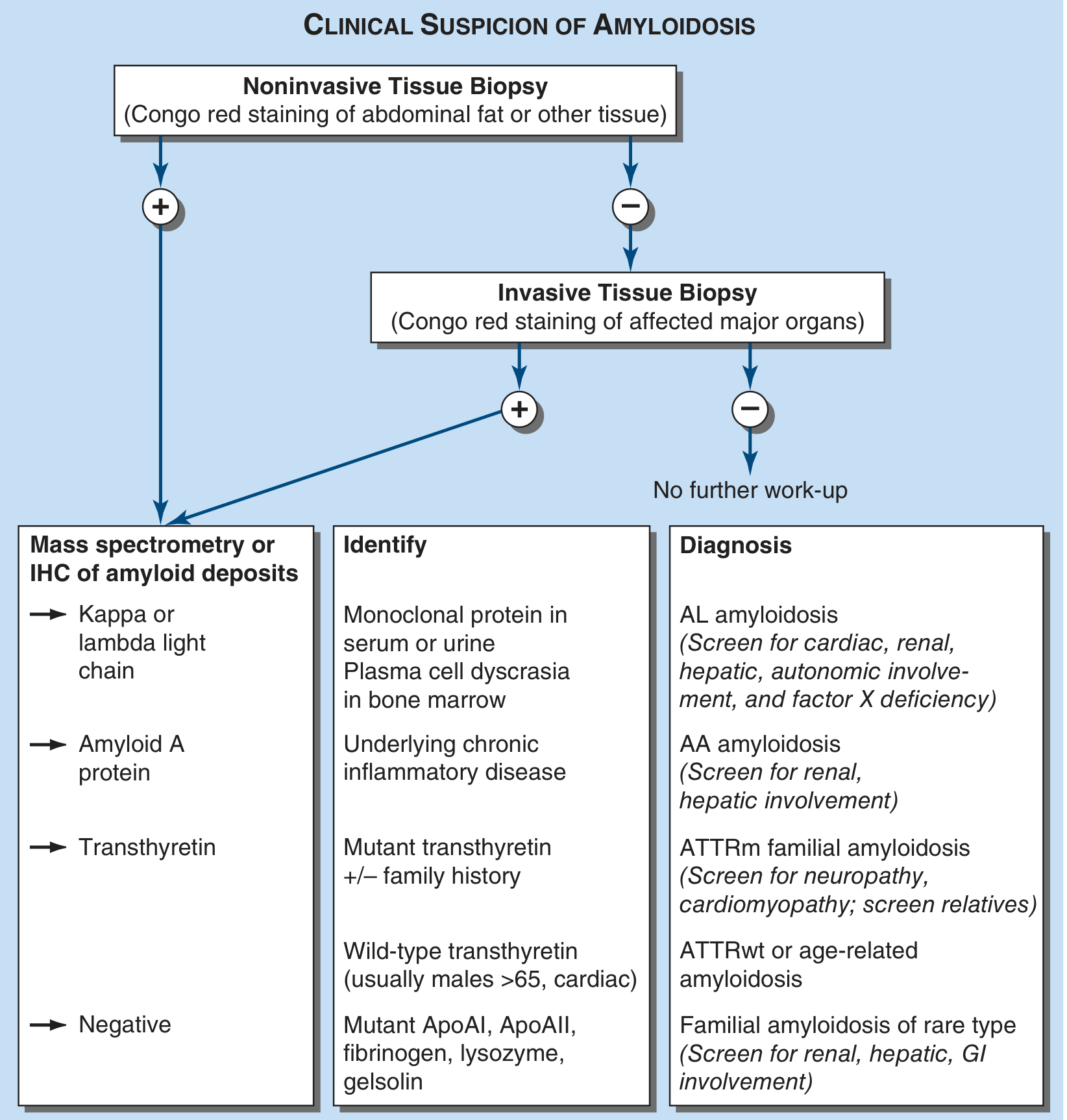
Fig. 117-1 from Harrison's Principles of Internal Medicine, 22E - Algorithm for the diagnosis of amyloidosis and determination of type.
Organ-Specific Consequences
| Organ | Pathophysiologic Effect |
|---|---|
| Kidney | Glomerular deposits → proteinuria, nephrotic syndrome, progressive CKD |
| Heart | Infiltrative/restrictive cardiomyopathy, diastolic dysfunction, elevated BNP/troponin, conduction abnormalities |
| Liver | Cholestasis (elevated ALP), hepatosplenomegaly; synthetic function usually preserved |
| Peripheral nerves | Length-dependent sensorimotor neuropathy, autonomic neuropathy (ATTR, AL) |
| GI tract | Motility disorders, malabsorption, bleeding (vascular amyloid) |
| Endocrine | Hypothyroidism, hypoadrenalism, hypopituitarism (AL amyloid in endocrine glands) |
| Soft tissue | Macroglossia, periorbital ecchymoses ("raccoon eyes"), carpal tunnel syndrome |
Sources
- Harrison's Principles of Internal Medicine, 22E (2025), Chapter 117 - Amyloidosis (primary reference)
- Fishman's Pulmonary Diseases and Disorders, 2-Volume Set - Chapter 63, Depositional Lung Diseases
- Rheumatology, 2-Volume Set (2022, Elsevier), Chapter 177 - AA Amyloidosis
This is a shared conversation. Sign in to Orris to start your own chat.