hyperkalemia
Reading File
Finding Sources
Searching PubMed
hyperkalemia management treatment
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, well-sourced content. Let me compile the full answer.
Hyperkalemia
Definition & Normal Range
Serum potassium > 5.0–5.5 mEq/L (normal 3.5–5.0 mEq/L). Levels ≥ 6.0 mEq/L are potentially dangerous; ≥ 7.0 mEq/L is a medical emergency.
Hyperkalemia is rare in healthy individuals (<1% of normal adults), underscoring the kidney's potent capacity for K⁺ excretion. Its presence in a non-acutely ill patient should prompt a search for impaired renal K⁺ handling.
Etiology
Causes are classified into three mechanisms:
1. Pseudohyperkalemia
Artifactual K⁺ release after phlebotomy — not true hyperkalemia.
- Hemolysis (most common — check plasma sample for pink discoloration)
- Prolonged tourniquet time or limb exercise with tourniquet
- Severe leukocytosis (WBC >70,000/mm³) or thrombocytosis (platelets 500–1000 × 10⁹/L — ~1/3 develop pseudohyperkalemia)
- Diagnose: serum [K⁺] > 0.3 mmol/L higher than simultaneous plasma [K⁺]
2. Transcellular Shift (ICF → ECF)
- Insulin deficiency / DKA — both hyperosmolarity and insulin deficiency drive K⁺ out of cells
- Metabolic acidosis — H⁺ exchange for intracellular K⁺
- Hyperosmolarity (e.g., mannitol, hyperglycemia)
- β-blockers — block β₂-mediated cellular K⁺ uptake
- Digoxin toxicity — blocks Na⁺/K⁺-ATPase
- Succinylcholine — depolarizing relaxant causes K⁺ efflux (dangerous in burns, denervation, rhabdomyolysis)
- Massive tissue necrosis / rhabdomyolysis — release of intracellular K⁺
3. Impaired Renal K⁺ Excretion (most common cause of chronic hyperkalemia)
| Category | Examples |
|---|---|
| CKD/ESKD | Reduced functioning nephrons → reduced distal K⁺ delivery and secretion |
| AKI (oliguric) | Reduced tubular flow |
| Hypoaldosteronism | Addison's, hyporeninemic hypoaldosteronism (type 4 RTA — DM, elderly, CKD) |
| Drugs | ACEi, ARBs, renin inhibitors, MR blockers (spironolactone, eplerenone, finerenone), K⁺-sparing diuretics (amiloride, triamterene), NSAIDs, heparin, calcineurin inhibitors (tacrolimus, cyclosporine), TMP-SMX |
| Hereditary | Pseudohypoaldosteronism type I (loss-of-function ENaC or MR mutations), PHA type II (WNK1/WNK4 mutations) |
| Tubulointerstitial disease | Amyloidosis, sickle cell, obstructive uropathy |
| Excessive intake | K⁺ supplements, salt substitutes (10–13 mmol K/g), enteral nutrition |
NSAIDs cause hyperkalemia by suppressing the renin-aldosterone axis and reducing GFR; COX-2-selective agents carry equal or greater risk. — Brenner & Rector's The Kidney
Clinical Manifestations
Hyperkalemia is often clinically silent until cardiac arrhythmia or arrest occurs — therefore every patient at risk must have an ECG.
Cardiac (primary danger)
ECG changes progress sequentially with rising K⁺ (Harrison's, 2025):
| Serum K⁺ (mEq/L) | ECG Finding |
|---|---|
| 5.5–6.5 | Peaked (tented) T waves, shortened QT interval |
| 6.5–7.5 | PR prolongation, P-wave flattening/loss |
| 7.0–8.0 | Widened QRS complex |
| > 8.0–9.0 | Loss of R-wave amplitude, ST depression |
| > 9.0 | Sine wave pattern → ventricular fibrillation → asystole |
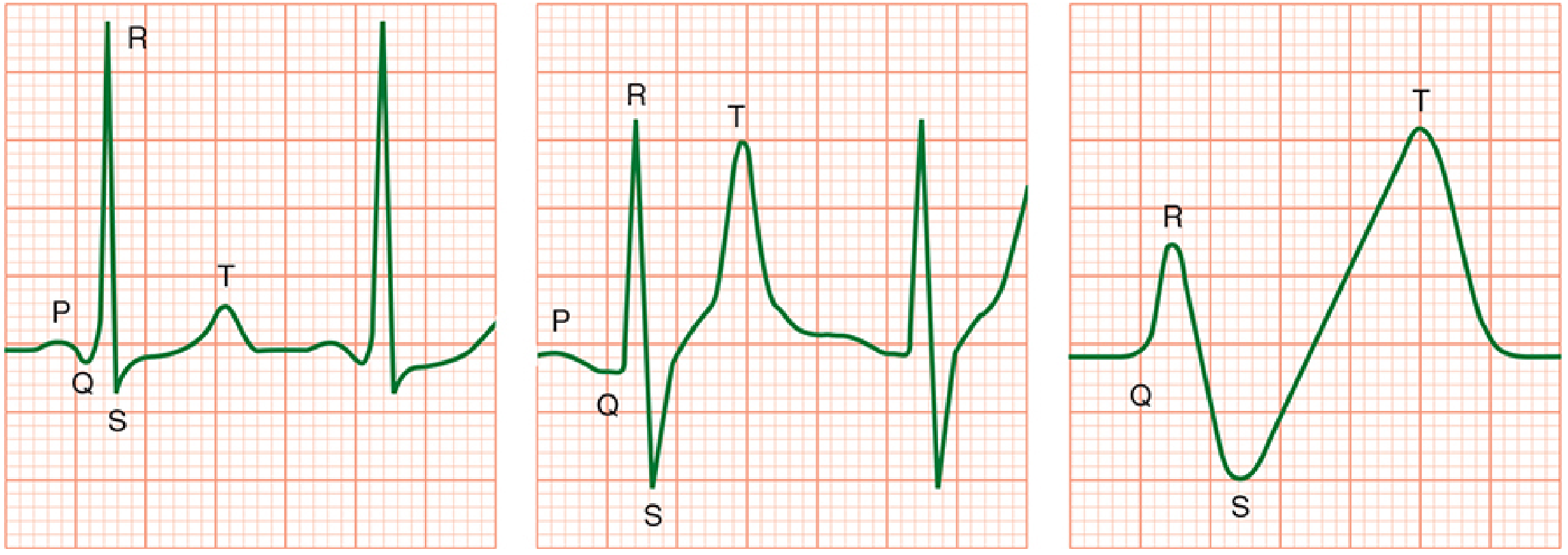
Contractility may be relatively preserved until late. Hypocalcemia, hyponatremia, and acidosis accentuate cardiac effects. ECG changes may be completely absent even when hyperkalemia is severe — a normal ECG does not exclude dangerous K⁺ elevation.
Neuromuscular
- Generalized skeletal muscle weakness (usually K⁺ > 8 mEq/L)
- Diaphragmatic weakness → respiratory failure in severe cases
- Ascending paralysis (rare)
Workup
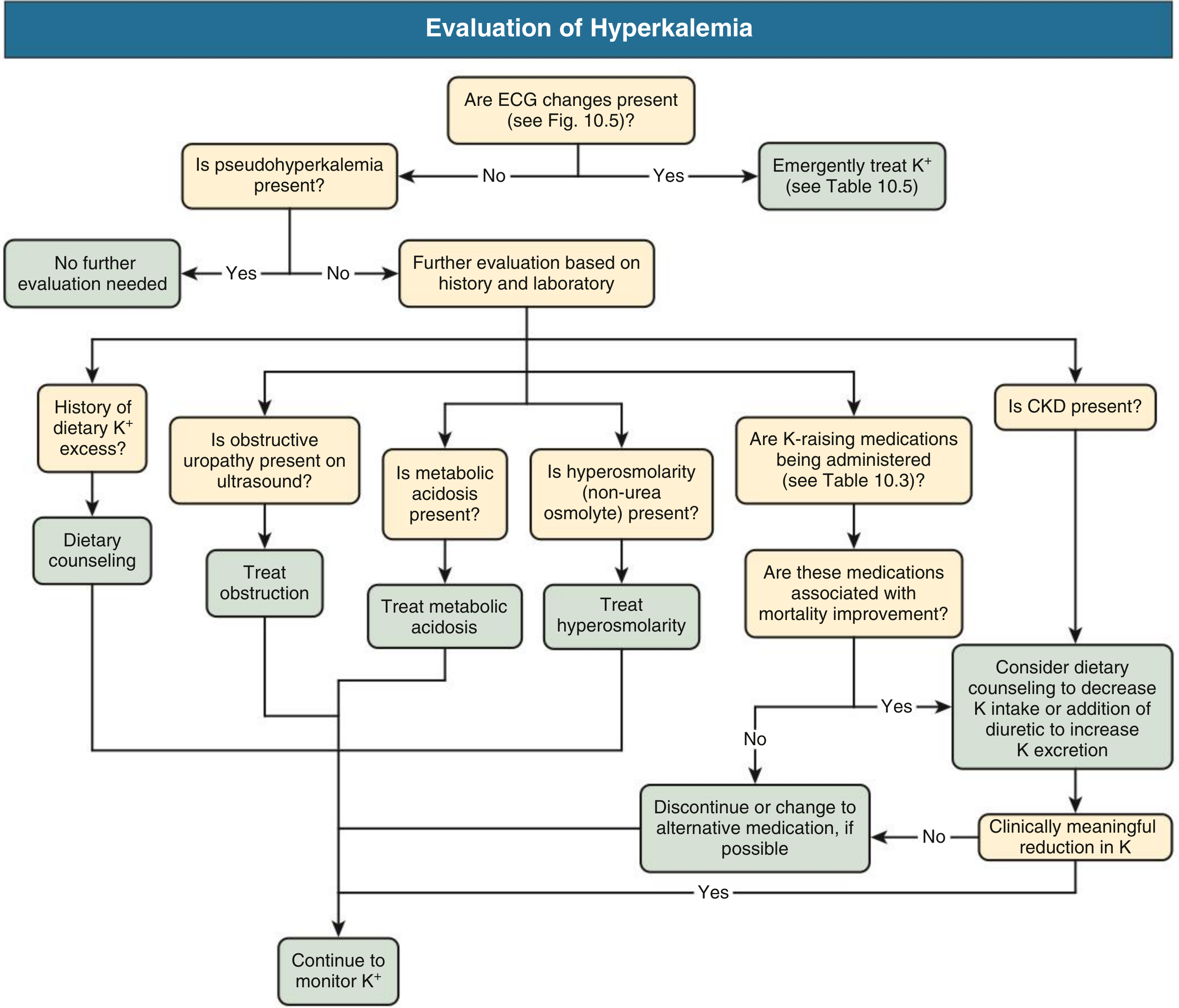
Key investigations:
- Repeat K⁺ in plasma (not serum) if hemolysis suspected
- ECG — immediately for any K⁺ ≥ 5.5 or clinical suspicion
- BMP: creatinine, BUN, glucose, bicarbonate, pH
- Urine K⁺, urine Cr, urine osmolality → calculate TTKG (transtubular K⁺ gradient)
- TTKG > 10 with hyperkalemia → extrarenal cause
- TTKG < 5–7 → aldosterone deficiency or resistance
- Aldosterone and plasma renin activity (PRA) if hypoaldosteronism suspected
- CBC (leukocytosis/thrombocytosis?)
Treatment
Treatment strategy follows three parallel goals:
1. Membrane Stabilization (immediate, minutes)
Calcium — antagonizes K⁺ effects on cardiac membrane directly; onset within 1–3 minutes, duration ~30–60 min.
- Calcium gluconate 10 mL of 10% IV over 2–3 minutes (or 5–10 mL); can repeat in 5 min if ECG unchanged
- Calcium chloride 3–5 mL of 10% IV (provides ~3× the calcium, use via central line)
- ⚠️ Caution in digoxin toxicity — calcium potentiates digoxin cardiotoxicity
2. Intracellular Shift (temporary — minutes to 1 hour)
| Agent | Dose | Onset | Duration | Notes |
|---|---|---|---|---|
| Insulin + glucose | 10 units regular IV + 25–50 g glucose (unless hyperglycemic) | 15–30 min | 4–6 h | Peak effect up to 1 h; monitor for hypoglycemia closely |
| Sodium bicarbonate | 1–2 mEq/kg IV | 15 min | 1–2 h | Most effective when metabolic acidosis present |
| β₂-agonists | Albuterol 10–20 mg nebulized (2–8× the bronchodilator dose!) or IV | 15–30 min | 2–4 h | ~25% non-responders; causes tachycardia |
Low-dose epinephrine infusion rapidly lowers K⁺ and provides inotropic support in severe/unstable cases (e.g., post-massive transfusion).
3. K⁺ Elimination (definitive)
| Method | When to use | Notes |
|---|---|---|
| Loop diuretics (furosemide) | Adequate renal function, no hypovolemia | Large doses may be needed in CKD |
| Potassium-binding resins | Chronic hyperkalemia, stable patients | |
| — Sodium polystyrene sulfonate (SPS/Kayexalate) | Oral or rectal | Safety and efficacy questioned; colonic necrosis risk |
| — Patiromer | Oral (preferred) | Better tolerated; 2025 meta-analysis supports use in CKD/HF to maintain RAAS inhibitor therapy |
| — Sodium zirconium cyclosilicate (ZS-9) | Oral, rapid onset | Acts within 1 hour |
| Dialysis | Life-threatening hyperkalemia, impaired renal function | Definitive; indicated when above measures fail or K⁺ > 6.5 with ECG changes |
4. Address the Underlying Cause
- Discontinue offending drugs (ACEi/ARB, K⁺-sparing diuretics, NSAIDs) if benefits don't outweigh risks
- Treat metabolic acidosis, obstructive uropathy
- Fludrocortisone for hypoaldosteronism
- Dietary K⁺ restriction counseling
Special Situations
- DKA: Serum K⁺ is often elevated at presentation despite total body K⁺ depletion — once insulin is started and acidosis corrects, K⁺ will drop rapidly. If K⁺ is already normal at presentation, defer insulin until K⁺ replacement is underway.
- CKD patients: Even modest K⁺ loads or drugs like β-blockers and ACEi can precipitate dangerous hyperkalemia. Any CKD patient in cardiac arrest should be presumed hyperkalemic.
- Succinylcholine: Potentially dangerous in pre-existing hyperkalemia (CKD, burns, denervation injury, rhabdomyolysis).
- RAAS inhibitors: Novel K⁺ binders (patiromer, ZS-9) now allow continuation of prognostically beneficial ACEi/ARB/MRB therapy in CKD/HF patients — a 2025 systematic review (PMID 40542996) confirms their efficacy in reducing hyperkalemia while optimizing RAAS inhibitor use.
Sources:
- Comprehensive Clinical Nephrology, 7th Ed., Ch. 10
- Harrison's Principles of Internal Medicine, 22E (2025)
- Morgan & Mikhail's Clinical Anesthesiology, 7e, Ch. 49
- ROSEN's Emergency Medicine, Ch. 83 & 114
- Brenner & Rector's The Kidney, 2-Volume Set, Ch. 13
This is a shared conversation. Sign in to Orris to start your own chat.