Classify photo dermatoses.Discuss PMLE in 10 marks format for 3rd yr dermatology post graduate theory examination
Reading File
Finding Sources
Finding Sources
Searching PubMed
polymorphic light eruption pathogenesis treatment
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Searching Images
polymorphic light eruption PMLE clinical morphology papules
I now have comprehensive material to write the complete answer. Let me compile the 10-mark PG examination format answer:
Classification of Photodermatoses & PMLE — 3rd Year Dermatology PG Theory
PART 1: CLASSIFICATION OF PHOTODERMATOSES
Photodermatoses are cutaneous disorders resulting from abnormal or expected reactions to ultraviolet (UV) radiation or visible light. They are classified as follows:
I. IDIOPATHIC (IMMUNOLOGICALLY-MEDIATED) PHOTODERMATOSES
(No exogenous photosensitizer; pathogenesis not fully known)
| Disorder | Key Feature |
|---|---|
| Polymorphous Light Eruption (PMLE) | Most common; papules/plaques in UV-exposed areas |
| Actinic Prurigo | Starts in childhood; cheilitis; HLA-DR4 association |
| Hydroa Vacciniforme | Children; vesicles → scarring; EBV association |
| Solar Urticaria | Urticarial wheals within minutes; IgE-mediated |
| Chronic Actinic Dermatitis (CAD) | Persistent eczema; broad-spectrum photosensitivity; elderly men |
II. PHOTOAGGRAVATED DERMATOSES
(Pre-existing conditions worsened by UV)
- Systemic Lupus Erythematosus (SLE) / Subacute LE / Discoid LE
- Rosacea
- Atopic dermatitis
- Seborrheic dermatitis
- Dermatomyositis
- Darier's disease
- Pemphigus
- Psoriasis (rarely)
III. CHEMICAL/DRUG-INDUCED PHOTOSENSITIVITY
A. Phototoxic reactions (dose-dependent, no sensitization required)
- Psoralens (psoralen + UVA = PUVA phototoxicity)
- Tetracyclines (especially doxycycline)
- NSAIDs (naproxen, piroxicam)
- Amiodarone, thiazides, fluoroquinolones, tar
B. Photoallergic reactions (immunologically mediated, T-cell)
- Sunscreen ingredients (benzophenones, PABA)
- Phenothiazines
- Sulfonamides
- Fragrances (musk ambrette)
IV. METABOLIC / GENODERMATOSES
A. Porphyrias
- Erythropoietic Protoporphyria (EPP) — most common porphyria with photosensitivity
- Porphyria Cutanea Tarda (PCT)
- Congenital Erythropoietic Porphyria (CEP / Günther disease)
B. DNA Repair Defect Disorders
- Xeroderma Pigmentosum (XP)
- Cockayne Syndrome
- Bloom Syndrome
- Trichothiodystrophy
C. Others
- Pellagra (niacin deficiency — Casal's necklace)
- Hartnup disease
PART 2: POLYMORPHOUS LIGHT ERUPTION (PMLE) — 10 MARKS
Definition & Introduction
Polymorphous Light Eruption (PMLE/PLE) is the most common idiopathic photodermatosis, characterized by recurrent, pruritic, polymorphic skin eruptions on sun-exposed areas, triggered by UV radiation, occurring predominantly in the spring and early summer in temperate climates. It was first described by Carl Rasch in 1900.
Epidemiology
- Prevalence is inversely related to latitude: highest in Scandinavia (~22%), moderate in UK and northern USA (10–15%), low in Australia (~5%), and rare in equatorial Singapore (~1%)
- This gradient reflects UV-induced immunologic tolerance ("hardening") from year-round sun exposure in sunny climates
- Female:Male ratio = 2:1 to 3:1
- Onset typically in the second and third decades of life; all races and skin phototypes affected
- Family history positive in 10–50% of cases
Pathogenesis
The exact mechanism is unknown. The leading hypothesis is:
Defective UV-induced immunosuppression / Delayed-type hypersensitivity (DTH) to a UV-induced neoantigen
- Normal UV exposure induces local immunosuppression (regulatory T cells, IL-10) — protecting against autoimmune reactions to UV-modified skin antigens
- In PMLE patients, this UV-induced immunosuppression is defective or absent → UV-modified skin proteins act as neoantigens → CD4⁺ T-cell mediated DTH reaction is mounted
- Reduced neutrophil infiltration after UV exposure has also been documented
- Action spectrum: UVB (290–320 nm) > UVA (320–400 nm); rarely visible light
- Hardening phenomenon: Repeated UV exposure progressively restores immunosuppression → lesions diminish as summer advances
Clinical Features
The morphology is polymorphous overall, but monomorphous in a given individual (lesion type stays constant for that patient).
Common morphological variants:
| Variant | Features |
|---|---|
| Papular / Erythematopapular | Most common; small erythematous papules |
| Papulovesicular | Vesicles superimposed on papules |
| Eczematous | Resembles contact dermatitis |
| Plaque-like | Indurated, fixed plaques; commoner in elderly; mimics lupus |
| Micropapular (pinpoint) | Common in darker skin phototypes (Fitzpatrick V–VI); mimics lichen nitidus |
| Erythematous | Diffuse redness |
Clinical variants:
- Benign Summer Light Eruption (BSLE): mild, diffuse variant with spontaneous clearing
- Juvenile Spring Eruption (JSE): occurs on the helices of ears in children (Fig. 3.23 — swollen, red ear helix)
Distribution:
- V-area of chest, neck, forearms, dorsum of hands, face
- Areas protected in winter (extensor forearms, chest V-area) are preferentially affected — reflecting the "change in UV dose" more than absolute amount
- Sun-protected areas (under chin, behind ears) are spared
Timing:
- Eruption appears 1–4 days after sun exposure (some report within 24 hours)
- Lesions last a few days to 1–2 weeks without scarring
- No scarring or atrophy (helps differentiate from LE and hydroa vacciniforme)
- Postinflammatory hyperpigmentation may occur in darker skin
Special point: PLE sine eruptione — pruritus only, without visible eruption; may develop full PLE later.
Histopathology
- Epidermis: Spongiosis (variable), may be normal
- Dermis: Dense, perivascular lymphocytic (CD4⁺) infiltrate in the superficial and deep dermis
- Striking papillary dermal edema — key early histological feature differentiating PMLE from LE
- No epidermal basal layer damage
- Direct Immunofluorescence (DIF): negative — important to distinguish from LE (DIF shows Ig/C3 deposits in LE)
Investigations / Evaluation
| Test | Finding/Purpose |
|---|---|
| Phototesting | UVB, UVA, sometimes visible light — reproduces lesions |
| Photoprovocation test | Repeated UV to simulate eruption; gold standard for diagnosis |
| Histopathology | Confirms diagnosis; excludes LE |
| ANA, anti-Ro/La, anti-dsDNA | To exclude SLE (10–20% of PMLE patients may have positive ANA) |
| Porphyrins (plasma/urine) | Exclude porphyria |
Note: 10–20% of PMLE patients may have positive ANA and family history of lupus; photosensitive SLE may present with PMLE-like eruptions years before SLE diagnosis — these patients must be followed up.
Differential Diagnosis
| Condition | Differentiating Feature |
|---|---|
| Lupus Erythematosus | DIF positive; ANA/anti-Ro positive; scarring; longer duration |
| Actinic Prurigo | Year-round; childhood onset; cheilitis; HLA-DR4 |
| Solar Urticaria | Urticarial wheals within minutes, not days; IgE-mediated |
| Photoallergic dermatitis | Photopatch test positive; requires prior sensitization |
| Phototoxic reaction | Drug history; onset within hours; burn-like |
| Chronic Actinic Dermatitis | Elderly men; persistent; low MED to broad spectrum |
| Erythropoietic Protoporphyria | Burning/stinging immediately; elevated protoporphyrins |
Treatment
A. Prophylactic / Preventive:
- Broad-spectrum sunscreens (SPF ≥ 30, UVA + UVB protection) — applied 20–30 min before sun exposure
- Protective clothing, sun avoidance (especially 10 AM–4 PM)
- Hardening/desensitization phototherapy (most effective long-term strategy):
- Narrowband UVB (NB-UVB, 311 nm) — preferred; 15–20 sessions in spring
- PUVA (psoralen + UVA) — for resistant cases
- These mimic the natural hardening process
B. Treatment of Acute Episodes:
- Topical corticosteroids — moderate-to-potent; for itching and inflammation
- Oral antihistamines — for pruritus relief
- Systemic corticosteroids (short course) — for severe, extensive flares
C. For Severe/Refractory Cases:
- Hydroxychloroquine (antimalarial) — especially for frequent, severe attacks
- Azathioprine — second-line immunosuppressant
- Beta-carotene — limited evidence
- Afamelanotide (α-MSH analogue) — stimulates melanin; used in clinical trials
Prognosis
- Spontaneous remission occurs in ~40% of patients over time (lesions become milder with advancing age)
- The hardening phenomenon means episodes are most severe in spring and become less severe as summer progresses
- Does not cause permanent scarring
- Small but important risk of later development of SLE — annual follow-up recommended
Clinical Images
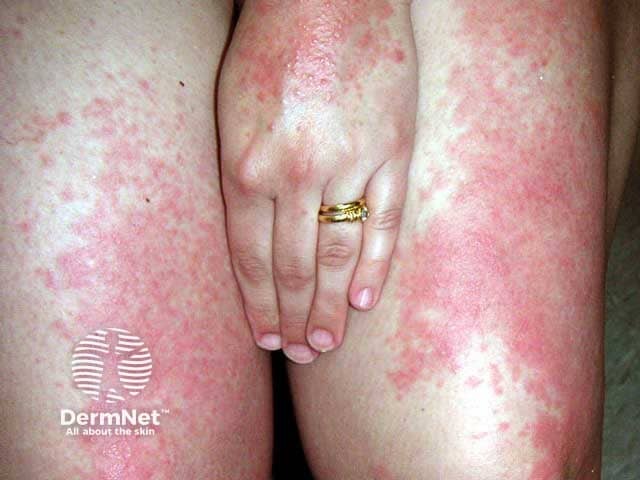
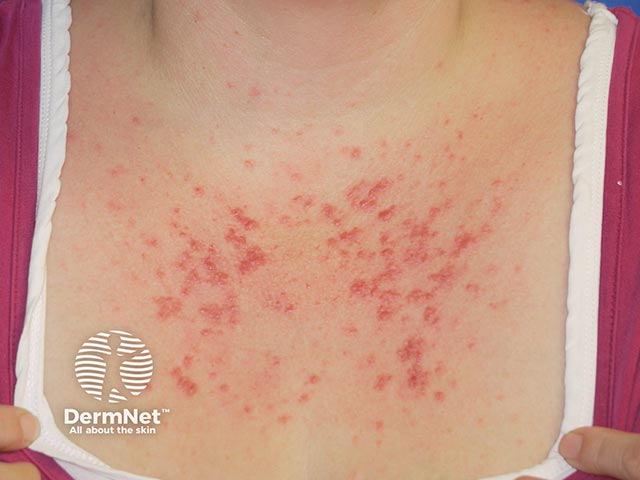
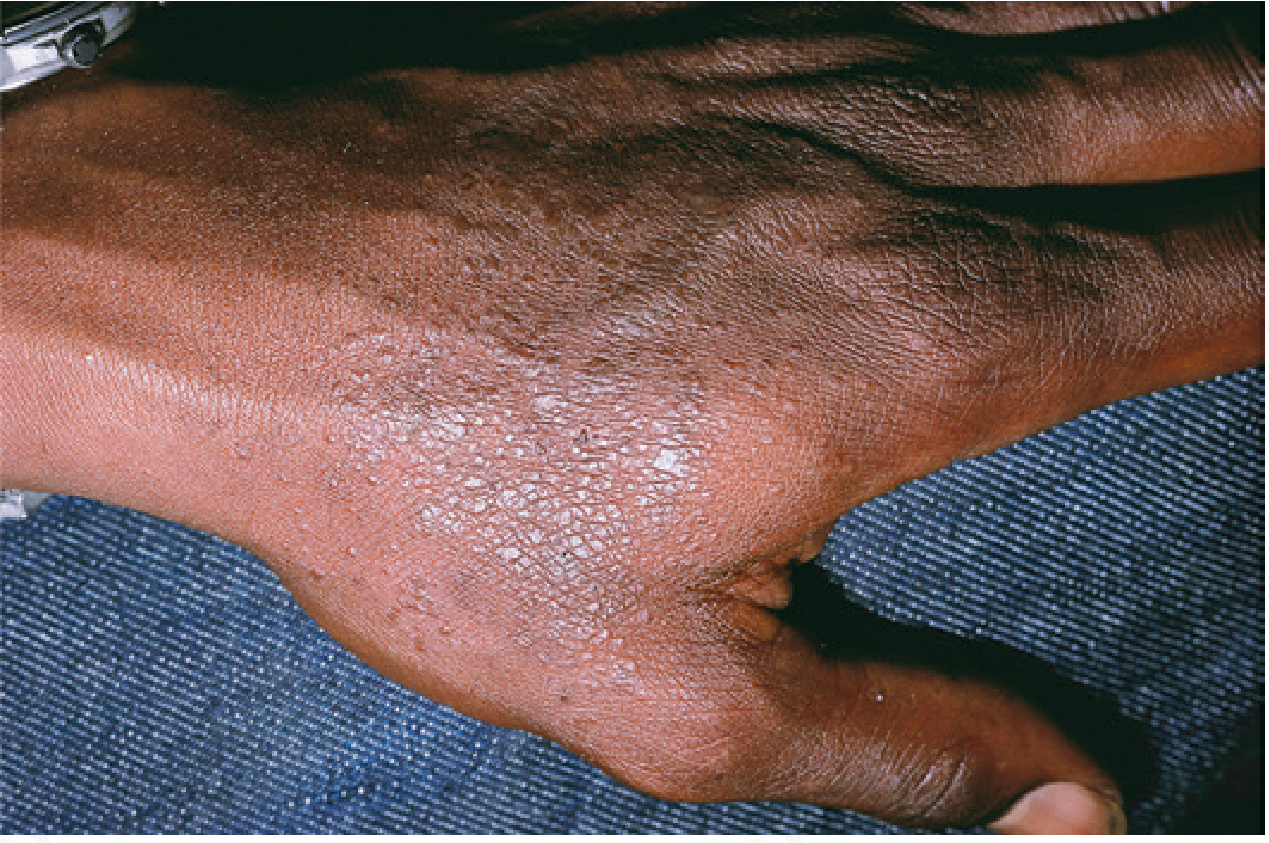
Summary Box (for quick revision)
| Feature | PMLE |
|---|---|
| Most common photodermatosis | ✓ |
| Action spectrum | UVB > UVA |
| Onset after sun | 1–4 days |
| Morphology | Papular (most common), vesicular, eczematous, plaque |
| DIF | Negative |
| Histology hallmark | Papillary dermal edema + lymphocytic infiltrate |
| Best prophylaxis | NB-UVB hardening |
| Distinguishing from LE | No scarring, DIF negative, no ANA in most |
References:
- Dermatology, 5th Edition (Bolognia, Schaffer, Cerroni) — Ch. 87 (Lim HW, Rosen CF)
- Andrews' Diseases of the Skin, 13th Ed — Ch. 3 (Idiopathic Photosensitivity Disorders)
- Fitzpatrick's Dermatology, Vol. 1–2
Classify annular erythema.Discuss EAC in 10marks format for 3rd yr dermatology post graduate theory examination
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
erythema annulare centrifugum trailing scale annular plaque
I now have comprehensive material from three authoritative dermatology textbooks plus clinical images. Let me compile the full answer:
Classification of Annular Erythemas & EAC — 3rd Year Dermatology PG Theory
PART 1: CLASSIFICATION OF ANNULAR ERYTHEMAS
Annular erythemas are a heterogeneous group of disorders characterized by erythematous lesions with annular, arciform, or polycyclic configurations. They are classified as follows:
I. FIGURATE (GYRATE) ERYTHEMAS — "The Classic Four"
| Disorder | Key Feature | Associated Condition |
|---|---|---|
| Erythema Annulare Centrifugum (EAC) | Trailing scale (inner border); slow centrifugal spread | Infections, malignancy, drugs, idiopathic |
| Erythema Marginatum | Rapidly migratory, evanescent, no scale | Acute rheumatic fever |
| Erythema Migrans | Expanding annular erythema at tick bite site | Lyme disease (Borrelia burgdorferi) |
| Erythema Gyratum Repens (EGR) | "Wood-grain" pattern; rapid migration (1 cm/day) | Internal malignancy (>70%; lung most common) |
II. INFECTIONS CAUSING ANNULAR ERYTHEMA
- Dermatophytosis (Tinea corporis) — leading scale (outer border); KOH positive
- Secondary syphilis — annular/psoriasiform plaques; serology positive
- Leprosy (Hansen disease) — hypopigmented anaesthetic annular plaques; AFB
- Lyme disease — erythema migrans (target lesion)
- Pityriasis rosea — herald patch + collarette scale
III. AUTOIMMUNE / CONNECTIVE TISSUE DISEASE
- Subacute Cutaneous Lupus Erythematosus (SCLE) — photodistributed; anti-Ro/SSA antibody
- Annular Erythema of Sjögren's Syndrome — associated with primary Sjögren's syndrome; anti-Ro/SSA positive; most common in Japanese
- Neonatal Lupus Erythematosus — transient annular plaques; maternal anti-Ro antibody
- Rowell Syndrome — SLE + EM-like annular lesions
IV. URTICARIAL / ALLERGIC ANNULAR ERYTHEMAS
- Urticaria — evanescent, lasts <24 h; wheals
- Erythema Multiforme — target lesions; acral > trunk
- Annular Urticaria
V. NEUTROPHILIC / EOSINOPHILIC DERMATOSES
- Eosinophilic Annular Erythema — recurrent annular plaques; eosinophilic infiltrate; may be associated with atopy
- Wells Syndrome (Eosinophilic Cellulitis) — "flame figures" on histology
VI. GRANULOMATOUS ANNULAR DERMATOSES
- Granuloma Annulare — skin-colored to erythematous papules in annular ring; no scale; palisading granulomas on histology
- Sarcoidosis — annular plaques; non-caseating granulomas
- Necrobiosis Lipoidica — pretibial, yellow-brown annular plaques
VII. DERMATOLOGICAL CONDITIONS WITH ANNULAR MORPHOLOGY
- Psoriasis — annular/gyrate variant; silvery scale; Auspitz sign
- Mycosis Fungoides (CTCL) — annular/poikilodermatous patches; cerebriform nuclei on histology
- Erythema Dyschromicum Perstans (Ashy Dermatosis) — grey annular macules
- Fixed Drug Eruption — recurring at same site; post-inflammatory pigmentation
PART 2: ERYTHEMA ANNULARE CENTRIFUGUM (EAC) — 10 MARKS
Definition
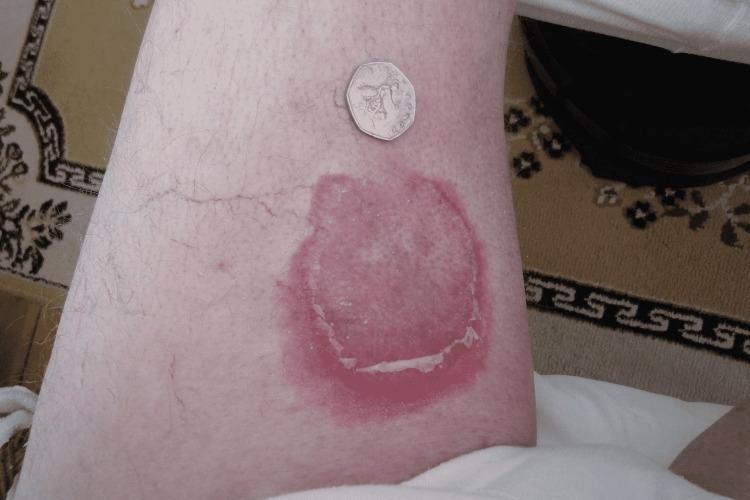
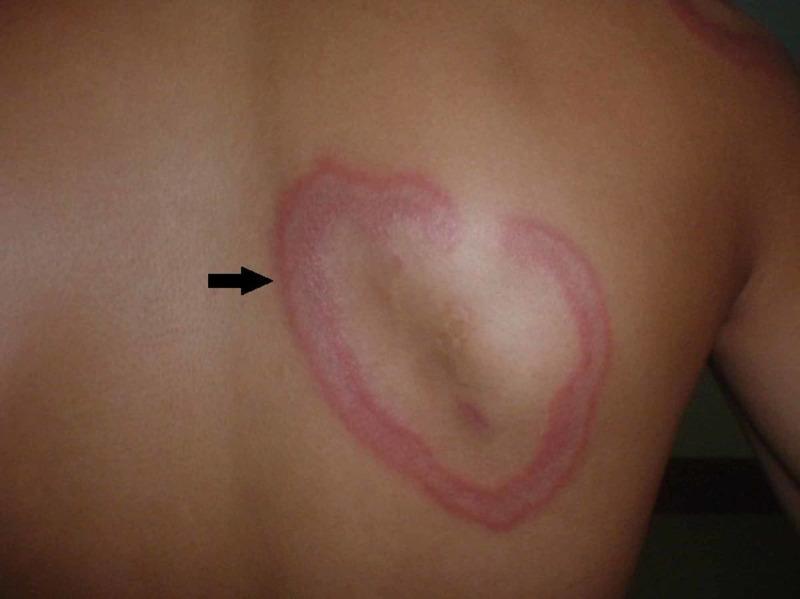
Erythema Annulare Centrifugum (EAC) is the most common figurate (gyrate) erythema, characterized by erythematous annular or polycyclic plaques that expand centrifugally with a pathognomonic trailing scale at the inner border, associated with central clearing. It is thought to represent a hypersensitivity reaction to various antigens.
Historical note: First described by Colcott-Fox in 1881 as "erythema gyratum perstans." The term EAC was coined by Jean Darier in 1916 to describe centrifugally expanding annular erythema with trailing scale.
Epidemiology
- Can occur in any age group; peak incidence in the 3rd–5th decade (Fitzpatrick's: 5th decade; Fitzpatrick's Ch. 46: 3rd–4th decade)
- No sex predilection (equal M:F)
- A rare autosomal dominant familial form ("familial annular erythema") exists
- Occurs in infants and children rarely
Etiology & Pathogenesis
EAC is best understood as a hypersensitivity/reactive inflammatory pattern triggered by various antigens. In up to 72% of cases, an associated condition can be found:
Infections (most common trigger):
- Dermatophytes — tinea pedis (48% in one series), tinea unguium
- Fungi — Candida, Penicillium (blue cheese ingestion)
- Viruses — EBV, VZV, poxvirus, HIV
- Bacteria — Pseudomonas, post-tonsillitis, Borrelia
- Parasites, ectoparasites (Phthirus pubis)
Drugs:
- Diuretics, NSAIDs, antimalarials, finasteride, amitriptyline
- Biologics: nivolumab, sorafenib, rituximab, ustekinumab, pegylated interferon-α + ribavirin
Malignancy (paraneoplastic EAC = PEACE):
- Lymphomas, leukemias, solid organ malignancies (13% in one series)
Others:
- Pregnancy, Crohn's disease, autoimmune endocrinopathies (Hashimoto's thyroiditis), hypereosinophilic syndrome
In one third of patients, no associated cause is found (idiopathic)
Mechanism: Peripheral migration of lesions reflects localized production of proinflammatory cytokines and vasoactive peptides — precise mechanism unknown. Superficial EAC likely represents a T-cell mediated hypersensitivity reaction causing epidermal spongiosis and scale.
Clinical Features
EAC presents as a pink papule that expands centrifugally forming an annular or polycyclic plaque with central clearing.
Rate of expansion: 2–3 mm/day; individual lesions can reach >6 cm diameter over 1–2 weeks.
Two Variants — Superficial vs. Deep
| Feature | Superficial EAC | Deep EAC |
|---|---|---|
| Border | Slightly elevated, erythematous | Indurated, firm, cord-like |
| Scale | "Trailing scale" at the inner margin ✓ | Absent (or minimal) |
| Pruritus | Common | Less common |
| Histology | Epidermal spongiosis + superficial perivascular infiltrate | Deep perivascular infiltrate; no epidermal changes |
| Associated trigger | Often infections (dermatophytes) | May be associated with systemic disease |
Key examination point: Scale in EAC is "trailing" (at the inner border, just behind the advancing edge) — this contrasts with tinea corporis where scale is "leading" (at the outer advancing border)
Distribution:
- Lower extremities (thighs, buttocks) — most common (~48%)
- Trunk (~28%)
- Upper extremities (~16%)
- Face — typically spared
Mucosal involvement: Absent
Course: Waxing and waning; individual lesions last days to months; total course may persist for months to years with eventual spontaneous remission.
Postinflammatory hyperpigmentation may occur; no scarring.
Histopathology
Superficial variant:
- Epidermis: focal spongiosis, parakeratosis, hyperkeratosis; ± basal vacuolar changes
- Dermis: dense perivascular lymphocytic infiltrate in the superficial dermis in a characteristic "coat-sleeve" arrangement around blood vessels
Deep variant:
- Epidermis: normal (no epidermal changes)
- Dermis: dense perivascular lymphocytic infiltrate concentrated in the upper and deep dermis — tight "coat-sleeve" arrangement
The "coat-sleeve" (sleeve-like) perivascular infiltrate is the histological hallmark of EAC — described in all major textbooks
Investigations
| Investigation | Purpose |
|---|---|
| KOH / Fungal culture | Exclude/confirm dermatophyte trigger (tinea pedis, tinea unguium) |
| Skin biopsy (histopathology) | Confirm EAC; distinguish superficial vs. deep; exclude mycosis fungoides |
| ANA, anti-Ro, anti-La | Exclude SCLE / Sjögren's associated annular erythema |
| CBC, LFT, serum LDH | Screen for associated malignancy/lymphoma |
| CT chest/abdomen | If malignancy suspected (paraneoplastic EAC) |
| RPR/VDRL | Exclude secondary syphilis |
| Dietary history (food diary) | Identify food triggers (blue cheese, tomatoes) |
| Drug review | Identify offending drug |
Differential Diagnosis
| Condition | Differentiating Features |
|---|---|
| Tinea corporis | Leading (outer border) scale; KOH/culture positive |
| Granuloma Annulare | Skin-colored/flesh-colored; no scale; palisading granulomas; no centrifugal migration |
| SCLE | Photodistributed; anti-Ro/SSA positive; DIF may show Ig deposits |
| Erythema Migrans | History of tick bite; single expanding lesion; responds to antibiotics |
| Erythema Marginatum | No scale; evanescent; child with rheumatic fever |
| Erythema Gyratum Repens | "Wood-grain/concentric rings" pattern; rapid migration (1 cm/day); malignancy |
| Annular Urticaria | Resolves <24 hours; wheals; dermographism |
| Mycosis Fungoides | Chronically persistent; cerebriform nuclei; T-cell clonality |
| Psoriasis (annular) | Silvery scale; Auspitz sign; nail changes |
| Secondary syphilis | Palmoplantar involvement; RPR positive |
Treatment
Step 1 — Treat the Underlying Cause:
- Identify and treat the trigger (e.g., antifungals for tinea pedis → often causes resolution of EAC)
- Withdraw offending drug
- Treat underlying malignancy (paraneoplastic EAC may resolve with treatment of cancer)
- Dietary modification if food trigger identified
Step 2 — Symptomatic Treatment:
| Agent | Use |
|---|---|
| Topical corticosteroids (moderate-to-potent) | First-line for symptomatic relief; reduces inflammation and pruritus |
| Topical calcipotriol (Vitamin D analogue) | Reported successful; useful as steroid-sparing agent |
| Antifungals (systemic or topical) | If dermatophyte trigger confirmed; often leads to complete resolution |
| Antihistamines | Pruritus relief |
For refractory/severe cases:
- Systemic corticosteroids — short courses
- Metronidazole — has been reported effective
- Erythromycin — reported benefit in some series
- Hydroxychloroquine, azathioprine, cyclosporine — anecdotal reports
Most cases eventually subside spontaneously, even without treatment, particularly if the trigger resolves.
Prognosis
- Chronic, waxing and waning course
- Individual lesions resolve in days to months
- Total disease duration: months to years (rarely persists permanently)
- Spontaneous remission is the eventual outcome in most patients
- Recurrence possible, especially if trigger (e.g., tinea) recurs
- No scarring; postinflammatory pigmentation may persist
Key Clinical Images
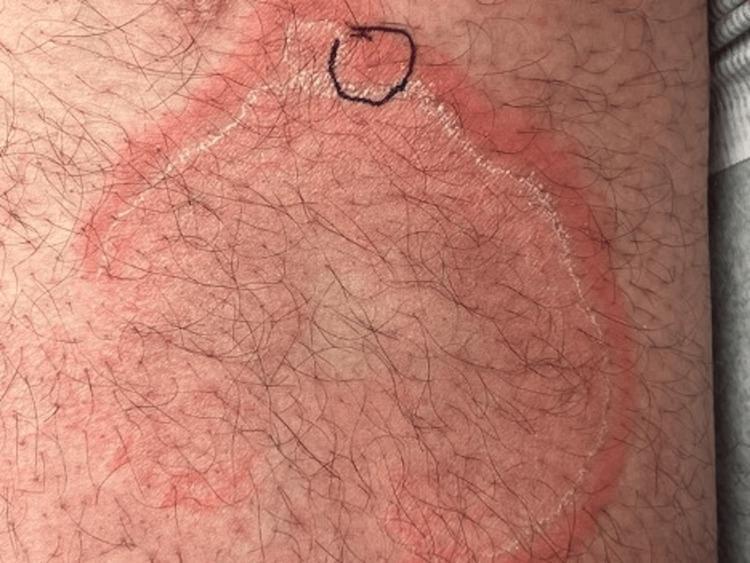
Summary Box — EAC at a Glance
| Feature | EAC |
|---|---|
| Most common figurate erythema | ✓ |
| Coined by | Darier, 1916 |
| Expansion rate | 2–3 mm/day |
| Hallmark sign | Trailing scale (inner border) |
| Histology | "Coat-sleeve" perivascular lymphocytic infiltrate |
| Most common trigger | Dermatophytosis (tinea pedis) |
| Paraneoplastic form | PEACE (Paraneoplastic EAC Eruption) |
| First-line Rx | Treat trigger + topical corticosteroids |
| Key D/D with tinea | Trailing scale (EAC) vs. leading scale (Tinea) |
References:
- Fitzpatrick's Dermatology, Vol. 1–2 — Ch. 46 (Ahn CS, Huang WW — EAC & Figurate Erythemas) and Table 19.1
- Dermatology, 5th Ed. (Bolognia, Schaffer, Cerroni) — Ch. 19 (Lim HW — Figurate Erythemas)
- Andrews' Diseases of the Skin, 13th Ed. — Ch. 7 (Gyrate Erythemas)
Tufted angioma in 5marks format for 3rd yr dermatology post graduate theory examination
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Searching Images
tufted angioma cannonball pattern histology vascular tufts
I have comprehensive content from three major dermatology textbooks. Here is the complete 5-mark PG answer:
TUFTED ANGIOMA — 5 Marks (3rd Year Dermatology PG Theory)
Definition
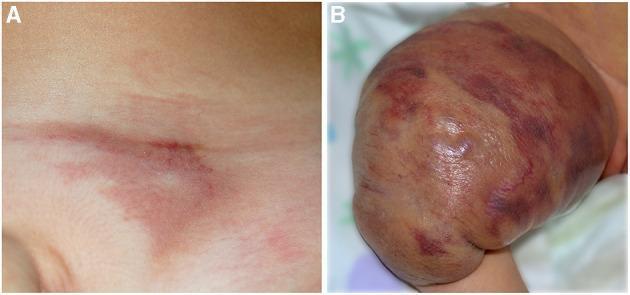
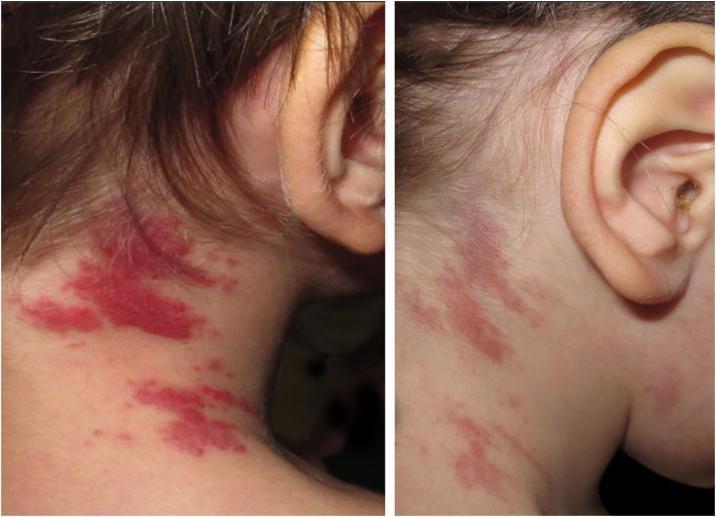
Tufted Angioma (TA) is a rare, benign vascular tumor of uncertain origin, characterized by slowly progressive mottled red plaques with superimposed angiomatous papules, caused by discrete tufts of tightly packed capillaries scattered in the dermis in a "cannonball" pattern on histology.
Synonyms: Angioblastoma of Nakagawa | Tufted hemangioma | Progressive capillary hemangioma
First described: Wilson-Jones and Orkin, 1989 (identical lesions were previously described as angioblastoma by Nakagawa, 1949)
Epidemiology
- Rare; no sex predilection
- ~25% are congenital; ~50% appear within the first year of life; over 50% present before age 5 years
- Adult onset described but uncommon
- Sporadic in the vast majority; rare familial cases reported
- Current consensus: TA represents a superficial, milder form of Kaposiform Hemangioendothelioma (KHE) on a shared histopathologic and molecular spectrum
Pathogenesis
- Etiology uncertain; based on immunostaining profile, a lymphatic endothelial origin is postulated
- TA and KHE share histologic and molecular overlap — considered part of a continuous spectrum
- Peripheral lesion growth reflects localized production of proinflammatory cytokines and vasoactive peptides
- Unlike infantile hemangioma, no GLUT-1 positivity; no known gender or gestational age correlates
- Platelet trapping within the tumor lobules leads to Kasabach-Merritt phenomenon in some cases
Clinical Features
Morphology:
- Begins as a subtle, poorly defined dull-red/violaceous macular stain that slowly thickens
- Evolves into a poorly defined, mottled red-dusky plaque (2–5 cm or larger) with superimposed angiomatous papules
- Can present as an exophytic, firm, rubbery violaceous nodule
- Associated features: overlying hypertrichosis (lanugo-like hair), hyperhidrosis, tenderness/pain (especially during KMP exacerbations)
Distribution:
- Most common: neck, upper trunk, shoulders
- Also: extremities, lower trunk; rarely face
Course:
- Slow centrifugal growth over months to years → eventual stabilization
- Rarely: complete spontaneous regression (more likely in congenital/early infantile lesions; 95% of regressing TAs do so within 2 years)
- Usually solitary; multifocal cases reported
Key complication — Kasabach-Merritt Phenomenon (KMP):
- Occurs more in congenital TAs and KHE
- Characterized by profound thrombocytopenia, microangiopathic hemolytic anemia, and consumptive coagulopathy due to platelet trapping within the tumor
- Life-threatening if untreated; elevated D-dimers, low fibrinogen
Histopathology — "Cannonball Pattern" (Hallmark)
- Multiple discrete lobules of tightly packed capillaries randomly scattered within the dermis and subcutis
- Lobules have a characteristic "cannonball" distribution — rounded clusters dispersed haphazardly through the dermis
- Capillary lumina are pinpoint-sized, occasionally containing fibrin microthrombi
- Lobules bulge into peripherally placed thin-walled lymphatic-like spaces (crescent-shaped spaces around tufts)
- Mitotic figures are rare
- Endothelial cells: may be focally spindled but less prominent than in KHE
- Dermal collagen between lobules may be normal or fibrotic
Immunohistochemistry:
| Marker | Tufted Angioma | Infantile Hemangioma | KHE |
|---|---|---|---|
| GLUT-1 | Negative | Positive | Negative |
| CD31 | Positive | Positive | Positive |
| CD34 | Positive (all endothelial cells) | + | Positive (luminal only) |
| Podoplanin / LYVE-1 / Prox-1 | Partially positive | Transient | Positive |
| SMA (smooth muscle actin) | Negative | — | — |
GLUT-1 negativity is the key differentiating feature from Infantile Hemangioma
Differential Diagnosis
| Condition | Differentiating Feature |
|---|---|
| Infantile Hemangioma | GLUT-1 positive; rapid growth then involution; no cannonball pattern |
| Kaposiform Hemangioendothelioma | Deeper, bulkier; more prominent spindle cells; more aggressive KMP |
| Kaposi Sarcoma | Slit-like spaces; spindle cell fascicles; plasma cells; HHV-8 positive; no tufting |
| Pyogenic Granuloma | Edematous stroma; granulation tissue; lobular but not cannonball |
| Vascular malformation | Present at birth; grows proportionally; no platelet trapping |
Clinical Images
Management
No universally accepted treatment guidelines exist. Therapy is guided by lesion size, activity, and presence of KMP.
| Scenario | Treatment |
|---|---|
| Small, flat, stable lesions | Observation ± low-dose aspirin (antiplatelet — with caution; stop around live vaccines in children to avoid Reye syndrome) |
| Larger growing lesions (without KMP) | Topical/intralesional corticosteroids, compression, pulsed dye laser, excision |
| Lesions with KMP | Systemic corticosteroids (rapidly effective) → Vincristine (classic second-line) → Sirolimus (proven highly effective) |
| Additional options | Propranolol, radiation, embolization (for KMP with KHE), IFN-α-2b |
| Aspirin maintenance | Used long-term after acute treatment to prevent platelet trapping recurrence |
Summary Box
| Feature | Tufted Angioma |
|---|---|
| Synonyms | Angioblastoma of Nakagawa |
| Peak onset | First year of life (congenital 25%) |
| Site | Neck, upper trunk |
| Hallmark morphology | Mottled red plaque + superimposed papules |
| Histology hallmark | Cannonball pattern of capillary tufts |
| Key differentiator from IH | GLUT-1 negative |
| Key complication | Kasabach-Merritt Phenomenon (KMP) |
| Relationship to KHE | Superficial, milder form of same spectrum |
| First-line for KMP | Sirolimus / systemic corticosteroids + vincristine |
References:
- Fitzpatrick's Dermatology, Vol. 1–2 — Ch. 118 (Tufted Angioma)
- Dermatology, 5th Ed. (Bolognia, Schaffer, Cerroni) — Ch. 114 (Lim HW/Rosen — Vascular Tumors)
- Andrews' Diseases of the Skin, 13th Ed. — Ch. 28
Pityriasis rubra Pilaris in 5marks format for 3rd yr dermatology post graduate theory examination
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.