Purine nucleotide degradation
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
Finding Sources
Reading File
I now have everything needed for a comprehensive answer. Let me compile it.
Purine Nucleotide Degradation
Purine nucleotides (AMP and GMP, derived from adenine and guanine) are ultimately degraded to uric acid in humans - the sole end product of purine catabolism. This distinguishes humans from most other mammals, which can further convert uric acid to allantoin via urate oxidase (uricase).
Overview: Where It Happens
| Source | Site of degradation | Fate |
|---|---|---|
| Dietary nucleic acids | Small intestine (enterocytes) | Converted to uric acid → blood → urine |
| Endogenous (de novo synthesis) | Primarily the liver | Free bases exported; salvaged by peripheral tissues |
| Dying cells (cell turnover) | Liver and peripheral tissues | Catabolized to uric acid |
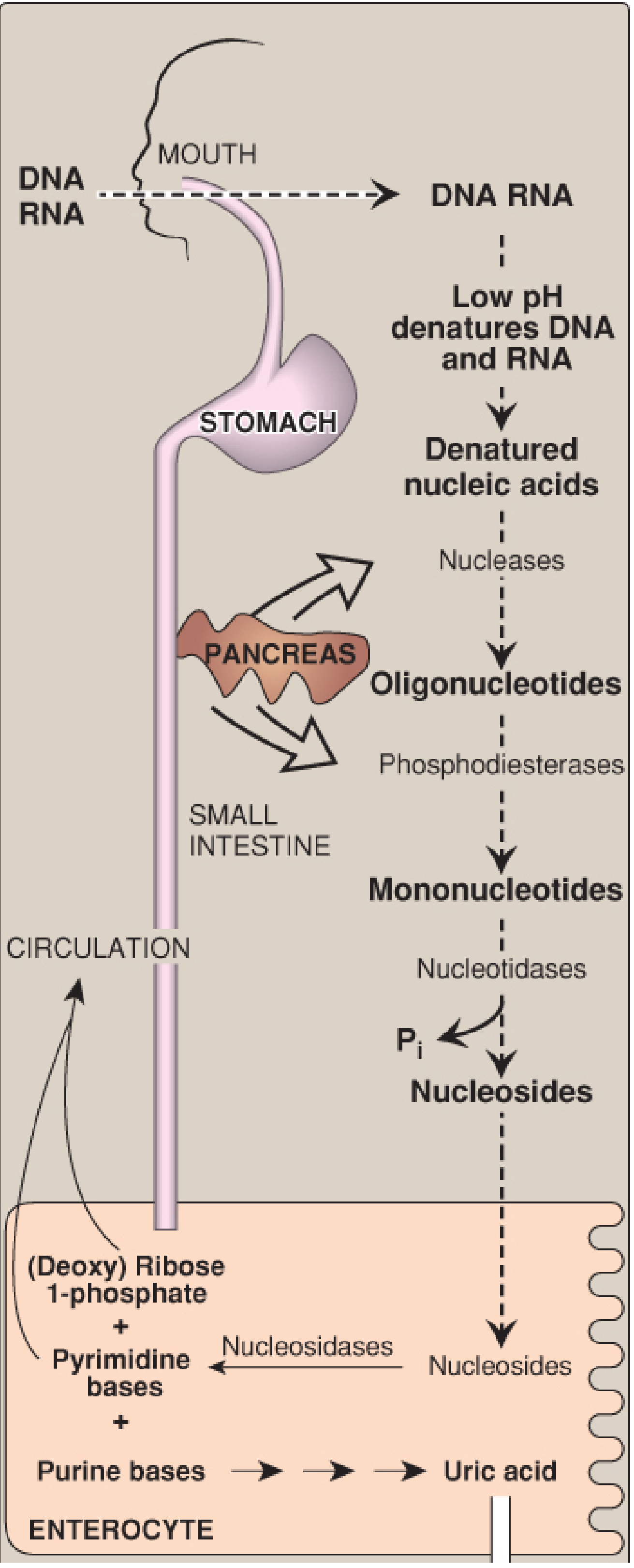
Pancreatic ribonucleases and deoxyribonucleases hydrolyze dietary RNA and DNA into oligonucleotides, then pancreatic phosphodiesterases produce 3'- and 5'-mononucleotides, and intestinal nucleotidases remove phosphate to yield nucleosides. These are absorbed by enterocytes via sodium-dependent transporters and cleaved by nucleosidases to free bases + (deoxy)ribose 1-phosphate.
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, p. 840
Stepwise Degradation to Uric Acid
The diagram below summarizes the full pathway:
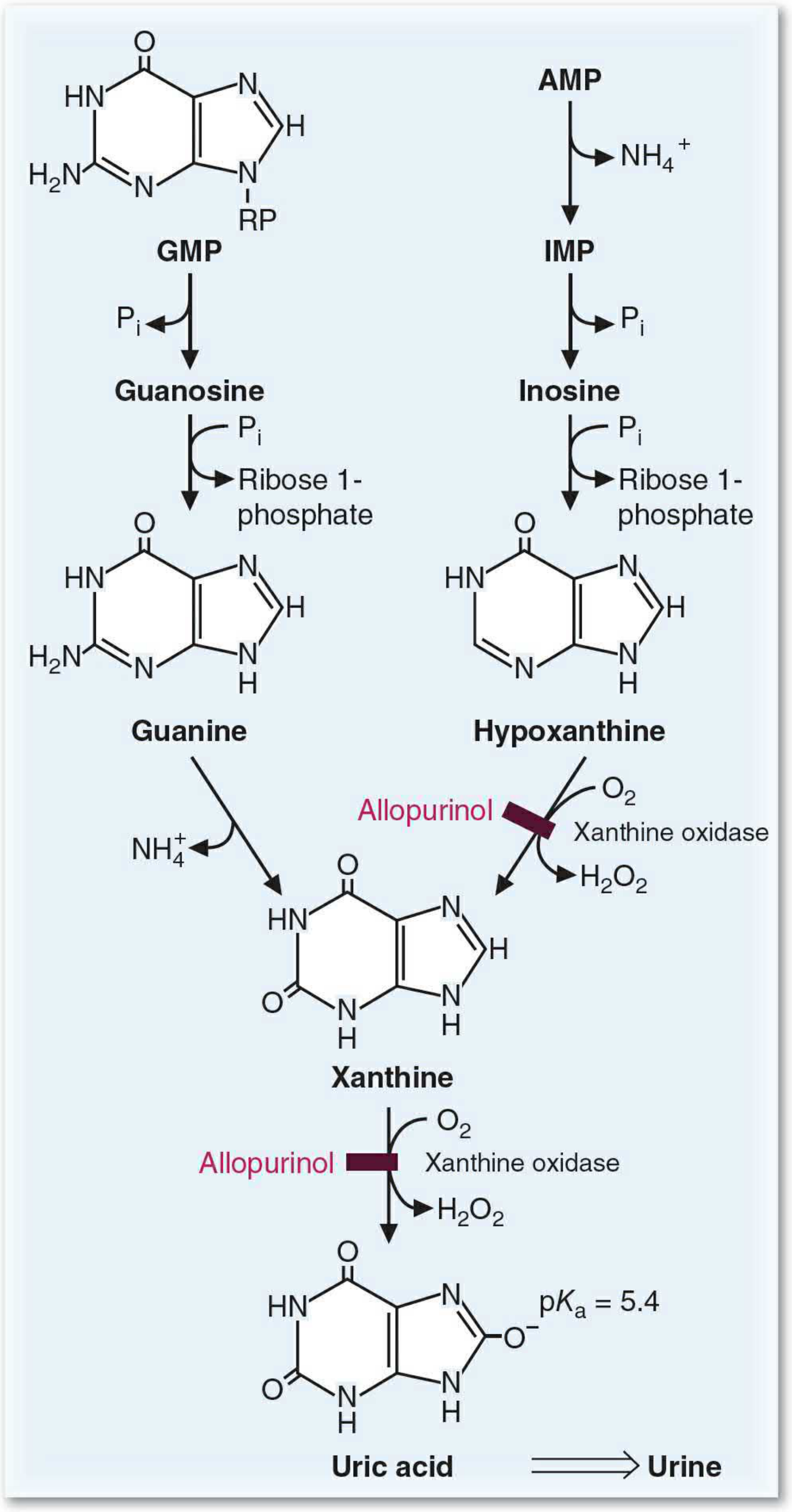
Step 1 - AMP to IMP (Deamination)
- AMP deaminase removes the amino group from AMP → IMP (inosine monophosphate), releasing NH4+
- Alternatively, adenosine deaminase (ADA) deaminates adenosine → inosine directly
Step 2 - Nucleotides to Nucleosides (Dephosphorylation)
- 5'-nucleotidase removes phosphate from IMP → inosine and from GMP → guanosine
Step 3 - Nucleosides to Free Bases (Phosphorolysis)
- Purine nucleoside phosphorylase (PNP) cleaves inosine → hypoxanthine + ribose 1-phosphate
- PNP cleaves guanosine → guanine + ribose 1-phosphate
- (A mutase interconverts ribose 1-phosphate and ribose 5-phosphate)
Step 4 - Guanine Deamination
- Guanine deaminase removes the amino group from guanine → xanthine + NH4+
Step 5 - Xanthine Oxidase (the key enzyme, x2 reactions)
- Xanthine oxidase (XO) oxidizes hypoxanthine → xanthine (uses O2, produces H2O2)
- XO further oxidizes xanthine → uric acid (uses O2, produces H2O2)
XO is a molybdenum-containing enzyme. It also exists in a form that uses NAD+ instead of O2 as the electron acceptor (xanthine dehydrogenase form).
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, p. 841; Basic Medical Biochemistry - A Clinical Approach - 6e, p. 1434
Dietary digestion overview
Properties of Uric Acid
- pKa = 5.4 → in the body (pH ~7.4), it is largely ionized as urate
- Urate is poorly soluble in aqueous environments; plasma urate is near its solubility limit
- In non-primate mammals, urate oxidase (uricase) converts uric acid to the more soluble allantoin
- Uric acid is also a potent antioxidant - one proposed reason humans retained it
Associated Diseases
1. Gout (hyperuricemia)
Gout occurs when uric acid levels exceed the solubility point, causing monosodium urate (MSU) crystal deposition in joints. The crystals trigger an inflammatory response (acute gouty arthritis), and chronic deposition causes tophi in soft tissues. Diagnosed by polarized light microscopy showing needle-shaped, negatively birefringent crystals in synovial fluid.
Causes of hyperuricemia:
- Underexcretion (>90% of cases) - primary or secondary (e.g., thiazide diuretics, lactic acidosis, lead exposure "saturnine gout")
- Overproduction - idiopathic; or from:
- Gain-of-function mutations in PRPP synthetase (increased PRPP → more de novo purines)
- Lesch-Nyhan syndrome (below)
- High cell turnover (myeloproliferative disorders, chemotherapy)
Treatment:
| Phase | Agent | Mechanism |
|---|---|---|
| Acute attack | Colchicine | Inhibits microtubule polymerization → impairs neutrophil migration |
| Acute attack | Indomethacin, prednisone | Anti-inflammatory |
| Chronic (underexcretors) | Probenecid, sulfinpyrazone | Uricosuric - increase renal uric acid excretion |
| Chronic (overproducers) | Allopurinol | Structural analog of hypoxanthine; inhibits XO |
| Chronic (refractory) | Febuxostat | Non-purine XO inhibitor |
Allopurinol mechanism: Oxidized to oxypurinol, a long-lived XO inhibitor → hypoxanthine and xanthine accumulate (more soluble than uric acid). In patients with normal HGPRT, the accumulated hypoxanthine is salvaged, reducing PRPP and de novo synthesis.
2. Lesch-Nyhan Syndrome (HGPRT deficiency)
- X-linked recessive deficiency of hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
- Cannot salvage hypoxanthine → IMP or guanine → GMP
- Hypoxanthine and guanine are degraded to uric acid instead → severe hyperuricemia
- Also causes self-mutilating behavior, choreoathetosis, spasticity, and intellectual disability (neurologic features dominate)
- PRPP accumulates (not consumed by salvage), driving excess de novo purine synthesis
3. Adenosine Deaminase (ADA) Deficiency
- ADA deaminates adenosine → inosine and deoxyadenosine → deoxyinosine
- Deficiency → toxic accumulation of dATP (deoxyadenosine triphosphate) in lymphocytes
- Inhibits ribonucleotide reductase → blocks DNA synthesis → lymphocyte death
- Leads to Severe Combined Immunodeficiency (SCID) - accounts for 10-20% of SCID cases
- Lymphocytes have the highest ADA activity of any tissue; they are most sensitive to its deficiency
4. Purine Nucleoside Phosphorylase (PNP) Deficiency
- Deficiency → accumulation of guanosine and inosine → toxic to T lymphocytes
- Results in T-cell immunodeficiency (predominantly T-cell SCID, B cells relatively spared)
Summary Pathway
AMP ──(AMP deaminase)──→ IMP
Adenosine ──(ADA)──→ Inosine
IMP ──(5'-nucleotidase)──→ Inosine ──(PNP)──→ Hypoxanthine + Ribose-1-P
GMP ──(5'-nucleotidase)──→ Guanosine ──(PNP)──→ Guanine + Ribose-1-P
Guanine ──(guanine deaminase)──→ Xanthine + NH4+
Hypoxanthine ──(Xanthine Oxidase)──→ Xanthine ──(Xanthine Oxidase)──→ URIC ACID
↑ [Allopurinol/Febuxostat block here]
Uric acid is excreted in the urine as the final end product.
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, p. 840-848; Basic Medical Biochemistry - A Clinical Approach - 6e, p. 1434-1435
Gout
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive data from Harrison's, Robbins, and Goodman & Gilman's. Let me compile the complete answer.
Gout
Gout is a crystal arthropathy caused by deposition of monosodium urate (MSU) crystals in joints and soft tissues, driven by chronic hyperuricemia. It is the most common inflammatory arthritis in adults and the only form of arthritis that humans develop spontaneously as a species - because unlike other mammals, humans lack uricase (urate oxidase), the enzyme that converts urate to the more soluble allantoin.
Pathogenesis
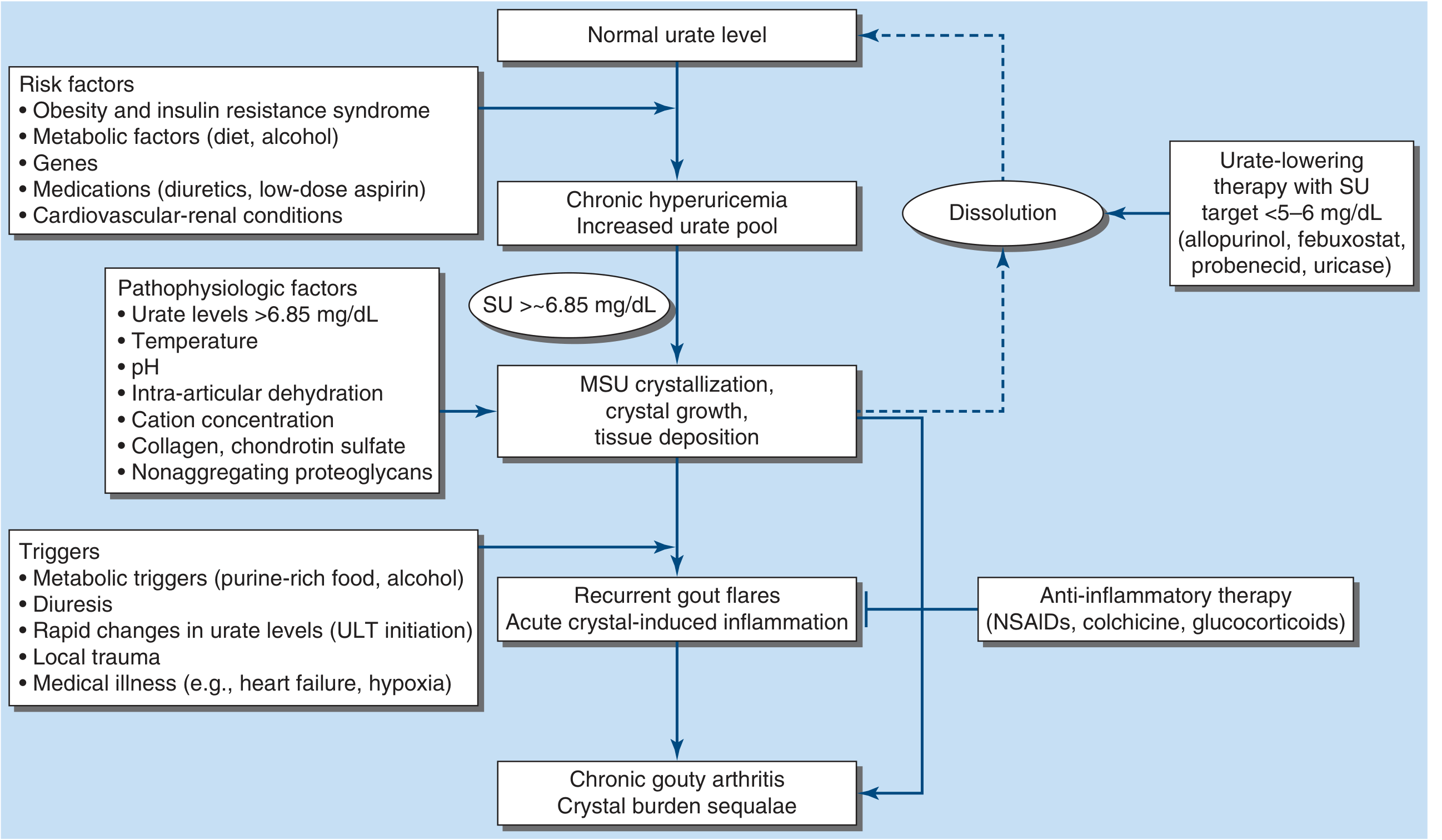
Hyperuricemia (plasma urate >6.8 mg/dL) is necessary but not sufficient - only ~10% of hyperuricemic individuals develop gout. The process unfolds in three stages:
1. Hyperuricemia
Uric acid levels reflect a balance of dietary intake, endogenous synthesis, and excretion. Hyperuricemia arises from:
| Mechanism | Frequency | Examples |
|---|---|---|
| Reduced excretion | ~90% of cases | Primary (idiopathic); diuretics; renal disease; lactic acidosis (lactate competes for urate excretion); lead nephropathy ("saturnine gout"); pyrazinamide |
| Overproduction | ~10% of cases | PRPP synthetase gain-of-function mutations; HGPRT deficiency (Lesch-Nyhan); myeloproliferative disorders; chemotherapy (tumor lysis); psoriasis |
Renal urate handling: Urate is filtered by the glomerulus, almost completely reabsorbed in the proximal tubule (via URAT1 and OAT10 transporters, sodium-dependent), then partially secreted. GLUT9 handles basolateral efflux. ABCG2 is a high-capacity urate efflux transporter in the gut and kidney; loss-of-function variants cause hyperuricemia.
At physiologic pH 7.4, uric acid (pKa 5.8) exists largely as urate - poorly soluble in aqueous environments. The saturation point is ~6.8 mg/dL; normal plasma urate is dangerously close to this limit.
2. MSU Crystal Formation & Inflammation
When urate supersaturates (>6.85 mg/dL), MSU crystals precipitate - favored by:
- Low temperature (explains predilection for peripheral, cooler joints - first MTP, ankles)
- Low pH
- Intra-articular dehydration
- Collagen and proteoglycans (promote crystal nucleation)
Crystal-induced inflammation cascade:
- MSU crystals are phagocytosed by resident synovial macrophages
- Crystals activate the NLRP3 inflammasome (NOD-like receptor protein 3)
- Inflammasome activates caspase-1 → cleaves pro-IL-1β → mature IL-1β
- IL-1β (and IL-6, IL-8, TNF-α) drives massive neutrophil recruitment
- Neutrophils phagocytose crystals → respiratory burst → lysosomal enzyme release, superoxide, leukotriene B4
- Crystals also physically damage phagolysosomal membranes → enzyme leakage
- Neutrophil mediators lower local pH → further urate precipitation (vicious cycle)
3. Chronic Tophaceous Gout
Without treatment, repeated flares → tophi - aggregates of MSU crystals surrounded by granulomatous inflammation (foreign body giant cells, macrophages, lymphocytes) in cartilage, tendons, and periarticular tissue. Growing crystal burden causes erosive joint destruction.
Clinical Stages
| Stage | Features |
|---|---|
| Asymptomatic hyperuricemia | Elevated urate, no symptoms; not treated as gout per se |
| Acute gout flare | Sudden-onset severe monoarthritis (or oligoarthritis); warm, red, exquisitely tender joint; often nocturnal onset; subsides spontaneously in 1-2 weeks |
| Intercritical gout | Asymptomatic intervals between flares |
| Chronic tophaceous gout | After years untreated; persistent synovitis, subcutaneous tophi, bone erosion, deformity |
Classic first presentation: Podagra - acute arthritis of the first metatarsophalangeal (MTP) joint, seen in 70-90% of gout patients. Also: tarsal joints, ankles, knees. Finger, wrist, and elbow joints are more common in elderly or advanced disease.
Demographics: Male:female = ~5:1 overall. Women represent only 5-20% of gout cases, typically postmenopausal, and often on diuretics.
Flare triggers: Purine-rich food, alcohol (especially beer and spirits), diuretic use, initiation of urate-lowering therapy (rapid urate changes loosen crystals from tophi), local trauma, medical illness (heart failure, respiratory illness/hypoxia), dehydration.
Risk Factors
- Older age, male sex
- Obesity, metabolic syndrome, insulin resistance
- Diet: red meat, shellfish/seafood, beer, spirits, high-fructose foods
- Medications: thiazide diuretics, loop diuretics, low-dose aspirin, pyrazinamide, ciclosporin
- Comorbidities: hypertension, renal disease, diabetes, hyperlipidemia, heart failure
- Genetic factors: HGPRT deficiency (partial = gout without neurology; complete = Lesch-Nyhan), PRPP synthetase superactivity, ABCG2/URAT1/SLC22A12 variants
Diagnosis
Gold standard: Needle aspiration of joint fluid (or tophus) + polarized light microscopy
| Crystal | Shape | Birefringence | Color under compensated polarized light |
|---|---|---|---|
| MSU (gout) | Needle- or rod-shaped | Strongly negative | Yellow when parallel to compensator axis (NYLOW: Negative Yellow pLarallel, blOW) |
| CPP (pseudogout) | Rhomboid, rod-shaped | Weakly positive | Blue when parallel to compensator axis |
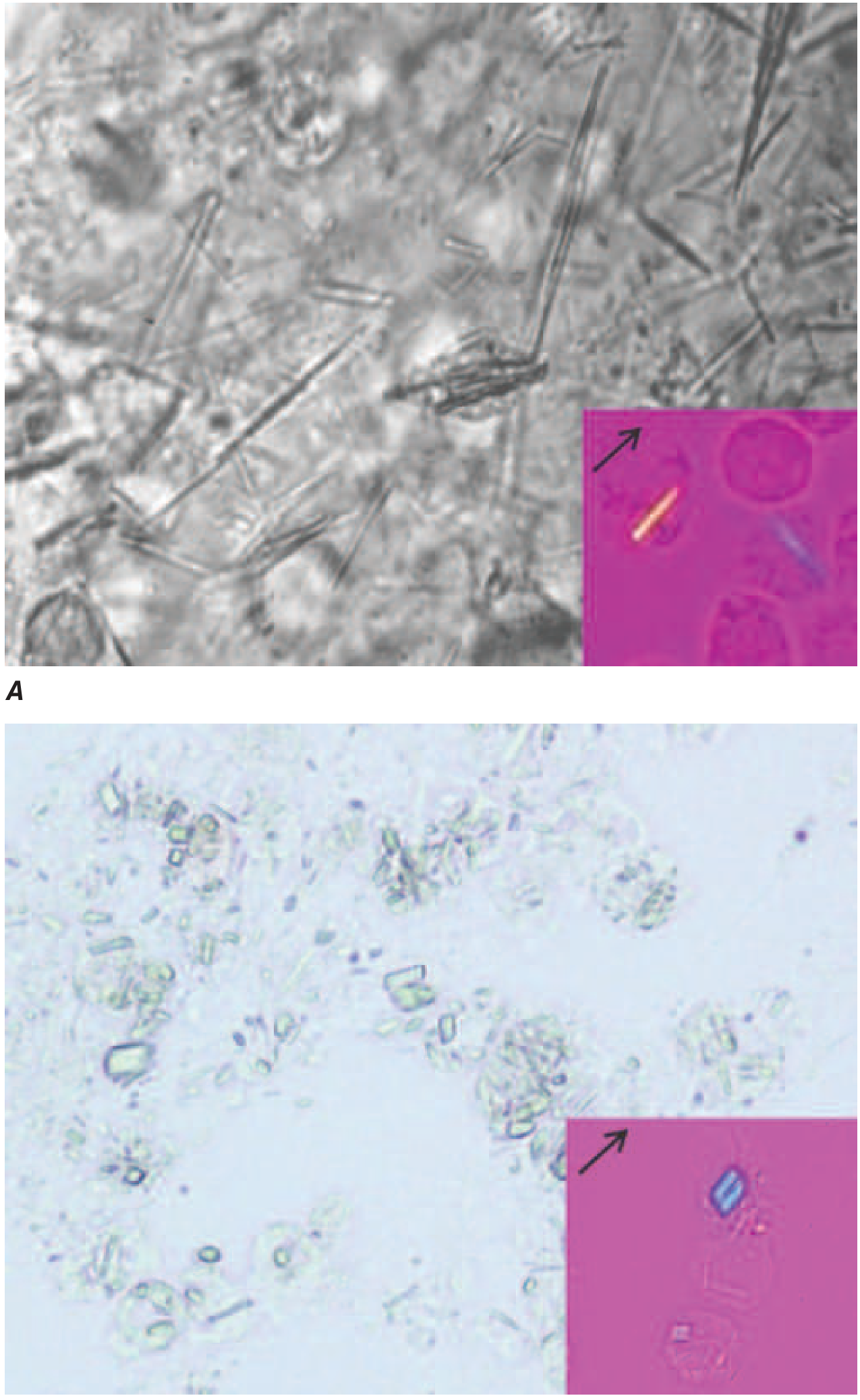
Synovial fluid WBC: typically 5,000-75,000/µL during acute flare (inflammatory pattern).
Key diagnostic caveat: Serum urate can be normal or low during an acute flare - inflammatory cytokines (especially IL-6) have uricosuric properties and can lower serum urate by ~2 mg/dL. Do not rule out gout based on a normal urate during a flare.
Imaging:
- Musculoskeletal ultrasound: "Double-contour sign" over articular cartilage (MSU deposition) - useful early
- Dual-energy CT (DECT): Identifies MSU based on chemical composition - highly specific
- Plain X-ray: In advanced disease - well-defined erosions with sclerotic margins and overhanging edges ("rat-bite" erosions), soft tissue tophi calcifications; typically absent early
Treatment
Acute Gout Flare
Ice packs and joint rest are helpful adjuncts. Pharmacologic options:
| Drug | Mechanism | Dose/Notes |
|---|---|---|
| Colchicine | Binds tubulin → prevents microtubule polymerization → impairs neutrophil migration; inhibits NLRP3 inflammasome → blocks IL-1β production | Low-dose: 1.2 mg at first sign of flare, then 0.6 mg in 1h. More effective and better tolerated than old high-dose regimens. Narrow therapeutic window. GI side effects. |
| NSAIDs (e.g., indomethacin, naproxen) | COX inhibition → ↓ prostaglandins | Most effective if started early. Avoid in renal impairment, heart failure, peptic ulcer disease. |
| Glucocorticoids (oral/IM/intra-articular) | Broad anti-inflammatory | Preferred in patients with contraindications to NSAIDs and colchicine; intra-articular injection for monoarthritis |
| Canakinumab | Monoclonal anti-IL-1β antibody | European approval for refractory cases; not FDA-approved for gout |
Note: Colchicine is eliminated via P-glycoprotein (MDR1) in the liver. Strong CYP3A4/P-gp inhibitors (cyclosporin, clarithromycin) significantly increase colchicine toxicity.
Urate-Lowering Therapy (ULT) - Chronic Management
Target: Serum urate <6 mg/dL (<360 µmol/L); <5 mg/dL (<297 µmol/L) in tophaceous gout, to drive crystal dissolution.
Important: Starting ULT can trigger acute flares (by mobilizing crystals from dissolving tophi). Prophylactic low-dose colchicine or NSAIDs should accompany ULT initiation for at least 3-6 months.
Xanthine Oxidase Inhibitors (XOI) - reduce urate synthesis
Allopurinol (first-line):
- Structural analog of hypoxanthine; oxidized by XO to oxypurinol, a long-lived (t½ ~18-30h) competitive inhibitor of XO
- Blocks both: hypoxanthine → xanthine AND xanthine → uric acid
- Accumulating hypoxanthine is salvaged by HGPRT → reduces PRPP → decreases de novo purine synthesis (bonus effect)
- Starting dose: 100 mg/day, titrate by 100 mg increments weekly; most patients: 300 mg/day
- Reduce dose in renal impairment
- Key drug interaction: Inhibits XO-mediated inactivation of azathioprine and 6-mercaptopurine → must reduce those doses to 25-33% when co-prescribed
- Adverse effects: Hypersensitivity rash (1-5%), rarely Stevens-Johnson syndrome / toxic epidermal necrolysis. Risk is strongly associated with HLA-B*5801 allele (screen in Korean, Thai, Han Chinese, Sardinian patients before initiating)
- Also inhibits warfarin metabolism (monitor INR)
Febuxostat (second-line):
- Non-purine XO inhibitor; inhibits both oxidized and reduced forms of XO (more complete inhibition than oxypurinol)
- Used when allopurinol is not tolerated or contraindicated
- Cardiovascular signal: higher rates of MI and stroke vs. allopurinol in cardiovascular-risk patients (CARES trial) - use with caution in existing CVD; prefer allopurinol when possible
Uricosuric Agents - increase renal urate excretion
| Drug | Mechanism | Notes |
|---|---|---|
| Probenecid | Blocks URAT1 → inhibits proximal tubule urate reabsorption | Used in underexcretors; avoid if GFR <30; ensure high urine output to prevent urate nephrolithiasis |
| Sulfinpyrazone | Uricosuric | Similar to probenecid |
| Benzbromarone | Potent uricosuric | Used in some countries; hepatotoxicity risk |
| Losartan, fenofibrate | Mild uricosuric (off-label) | Secondary benefits in comorbid hypertension/hyperlipidemia |
Uricase Agents - enzymatic urate degradation
| Drug | Notes |
|---|---|
| Pegloticase | Recombinant PEGylated uricase; converts urate → allantoin. For refractory chronic tophaceous gout. IV infusions. Infusion reactions; develops neutralizing antibodies. |
| Rasburicase | Recombinant uricase; used for tumor lysis syndrome (not standard chronic gout therapy). Contraindicated in G6PD deficiency (generates H2O2). |
Complications
| Complication | Mechanism |
|---|---|
| Chronic gouty arthritis | Erosive joint destruction from growing tophaceous deposits |
| Tophaceous deposits | In pinna of ear, olecranon bursa, Achilles tendon, finger pads |
| Uric acid nephrolithiasis | Urate stones (radiolucent on plain X-ray); acidic urine promotes stone formation |
| Urate nephropathy | MSU crystal deposition in renal interstitium → CKD |
| Acute uric acid nephropathy | Massive urate precipitation in renal tubules (tumor lysis) → acute kidney injury |
Differential Diagnosis
Gout must be distinguished from:
- Pseudogout (CPPD) - calcium pyrophosphate crystals, weakly positive birefringence, rhomboid shape; often affects knees and wrists
- Septic arthritis - always culture synovial fluid (infection can coexist with MSU crystals)
- Cellulitis - gout over first MTP can mimic cellulitis ("pseudocellulitis")
- Reactive arthritis, psoriatic arthritis - can overlap
Lifestyle & Dietary Advice
-
Avoid purine-rich foods: organ meats, red meat, shellfish (especially mussels, scallops, anchovies)
-
Avoid beer and spirits; moderate wine consumption is less strongly associated with gout
-
Avoid high-fructose corn syrup (fructose promotes purine synthesis and reduces urate excretion)
-
Low-fat dairy products are associated with decreased gout risk
-
Maintain high fluid intake (>2L/day)
-
Weight loss, treat metabolic syndrome, discontinue offending drugs if possible
-
Harrison's Principles of Internal Medicine 22E, p. 3000-3003; Robbins & Kumar Basic Pathology, p. 840-846; Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 867-875
Pyrimidine synthesis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I have everything needed. Here is the comprehensive answer.
Pyrimidine Synthesis
Key Distinction from Purine Synthesis
| Feature | Pyrimidine synthesis | Purine synthesis |
|---|---|---|
| Ring assembly | Ring built first, then attached to ribose | Ring assembled directly on pre-existing ribose (PRPP) |
| Location | Cytosol (first 3 steps); inner mitochondrial membrane (step 4) | Cytosol |
| End product of ring | Free pyrimidine base (orotate) → then nucleotide | Nucleotide (IMP) from the outset |
Atomic Origins of the Pyrimidine Ring
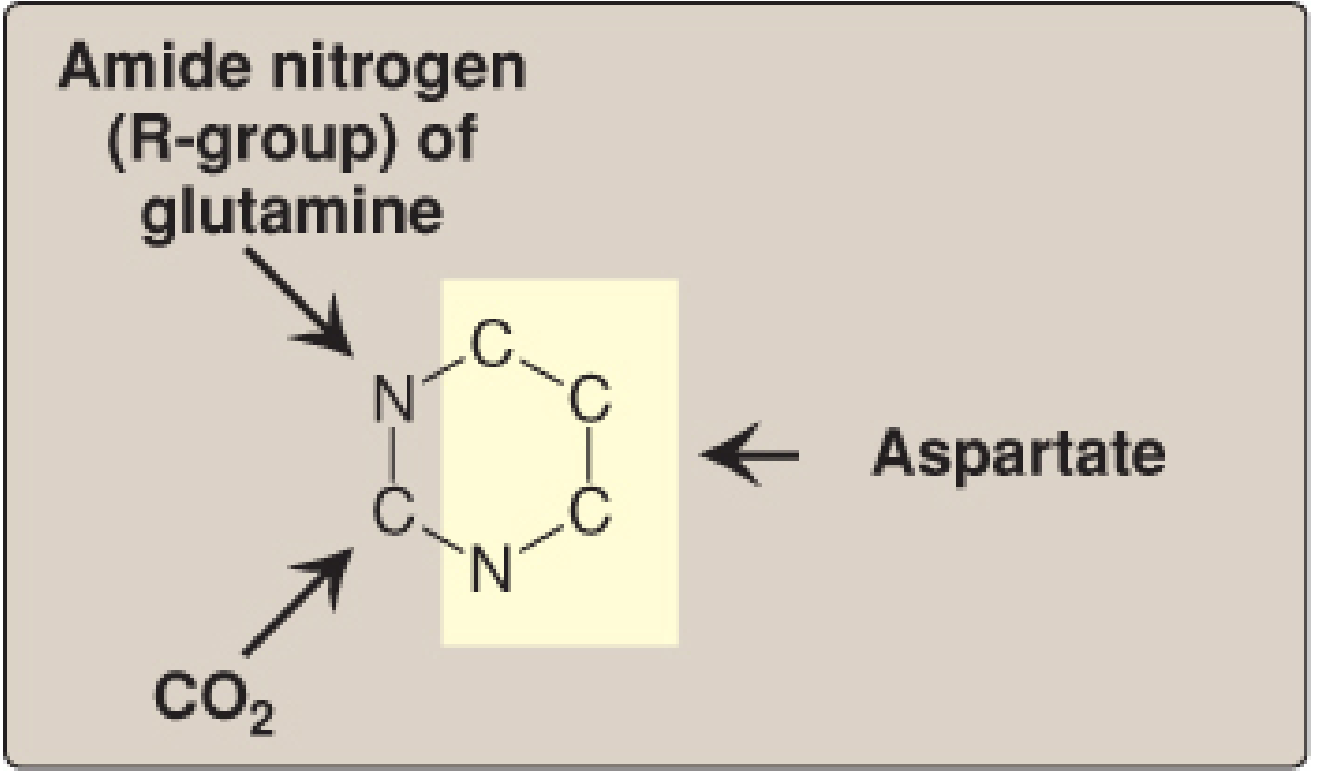
- N-1 and C-2: from carbamoyl phosphate (derived from glutamine's amide nitrogen + CO₂)
- N-3, C-4, C-5, C-6: from the entire aspartate molecule
Stage 1: De Novo Synthesis of the Pyrimidine Ring (Steps 1-3)
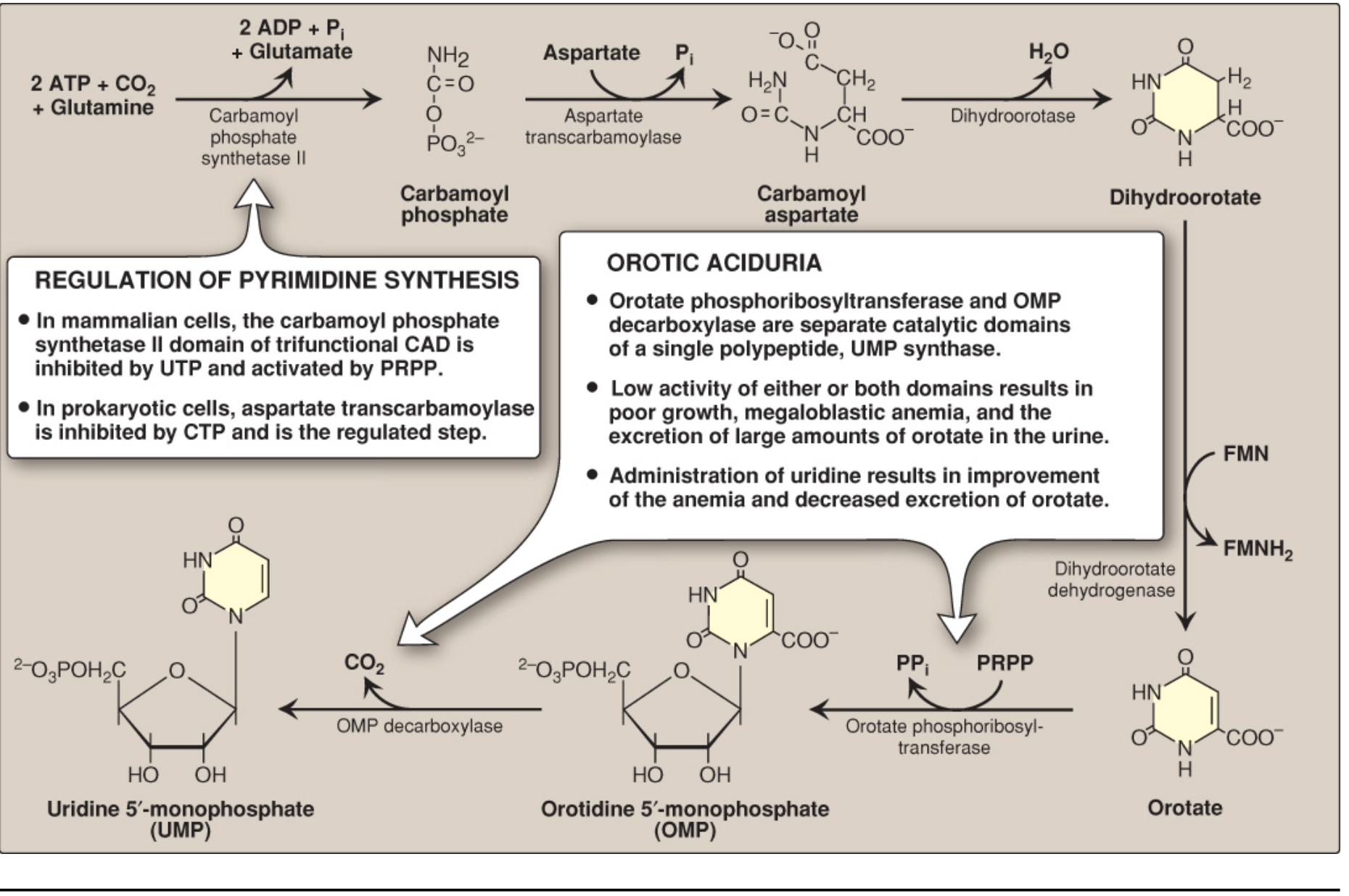
In mammals, the first three enzymes of the pathway are encoded as a single trifunctional polypeptide called CAD (Carbamoyl phosphate synthetase II - Aspartate transcarbamoylase - Dihydroorotase).
Step 1 - Carbamoyl phosphate synthesis (RATE-LIMITING STEP)
Enzyme: Carbamoyl phosphate synthetase II (CPS II)
Location: Cytosol
Reaction:
Glutamine + CO₂ + 2 ATP → Carbamoyl phosphate + Glutamate + 2 ADP + Pi
CPS II vs CPS I comparison:
| Feature | CPS I (urea cycle) | CPS II (pyrimidine synthesis) |
|---|---|---|
| Location | Mitochondria | Cytosol |
| Nitrogen source | Ammonia (NH₄⁺) | γ-Amide of glutamine |
| Activator | N-acetylglutamate | PRPP |
| Inhibitor | - | UTP (feedback inhibition) |
Step 2 - Carbamoylaspartate formation
Enzyme: Aspartate transcarbamoylase (ATCase)
Carbamoyl phosphate + Aspartate → Carbamoylaspartate + Pi
(Note: In bacteria/prokaryotes, ATCase is the regulated step, inhibited by CTP and activated by ATP - not in mammals)
Step 3 - Ring closure
Enzyme: Dihydroorotase
Carbamoylaspartate → Dihydroorotate + H₂O
Intramolecular condensation closes the 6-membered ring.
Stage 2: Orotate Formation and Attachment to Ribose (Steps 4-6)
Step 4 - Oxidation to orotate
Enzyme: Dihydroorotate dehydrogenase (DHODH)
Location: Inner mitochondrial membrane (only step outside the cytosol)
Dihydroorotate + FMN → Orotate + FMNH₂
FMN is reduced; electrons eventually transferred to ubiquinone in the respiratory chain.
Step 5 - Attachment to ribose (PRPP donation)
Enzyme: Orotate phosphoribosyltransferase
Location: Cytosol (part of UMP synthase bifunctional enzyme)
Orotate + PRPP → Orotidine 5'-monophosphate (OMP) + PPi
Pyrophosphate (PPi) release makes this reaction thermodynamically irreversible (pulled forward by pyrophosphatase). This is the step where ribose is attached.
Step 6 - Decarboxylation to UMP
Enzyme: OMP decarboxylase (orotidylic acid decarboxylase)
Location: Cytosol (part of UMP synthase)
OMP → Uridine monophosphate (UMP) + CO₂
Steps 5 and 6 are carried out by a single bifunctional enzyme: UMP synthase.
Full De Novo Pathway Diagram
Stage 3: UMP → All Other Pyrimidine Nucleotides
UMP → (UMP kinase) → UDP → (NDPK) → UTP
CTP Synthesis
Enzyme: CTP synthetase
UTP + Glutamine + ATP → CTP + Glutamate + ADP + Pi
- Glutamine provides the amino group added at C-4 of uracil to form cytosine
- Note: This amination can only occur at the triphosphate level (not at UMP or UDP)
- CTP is a feedback inhibitor of CTP synthetase (end-product inhibition)
Deoxyribonucleotides
- UDP and CDP are substrates for ribonucleotide reductase → dUDP and dCDP
- dCDP → dCTP (for DNA) or dephosphorylated → dCMP → deaminated → dUMP
- dUDP → phosphorylated → dUTP → rapidly hydrolyzed by dUTPase to dUMP (prevents erroneous U incorporation into DNA)
dTMP Synthesis (Critical for DNA)
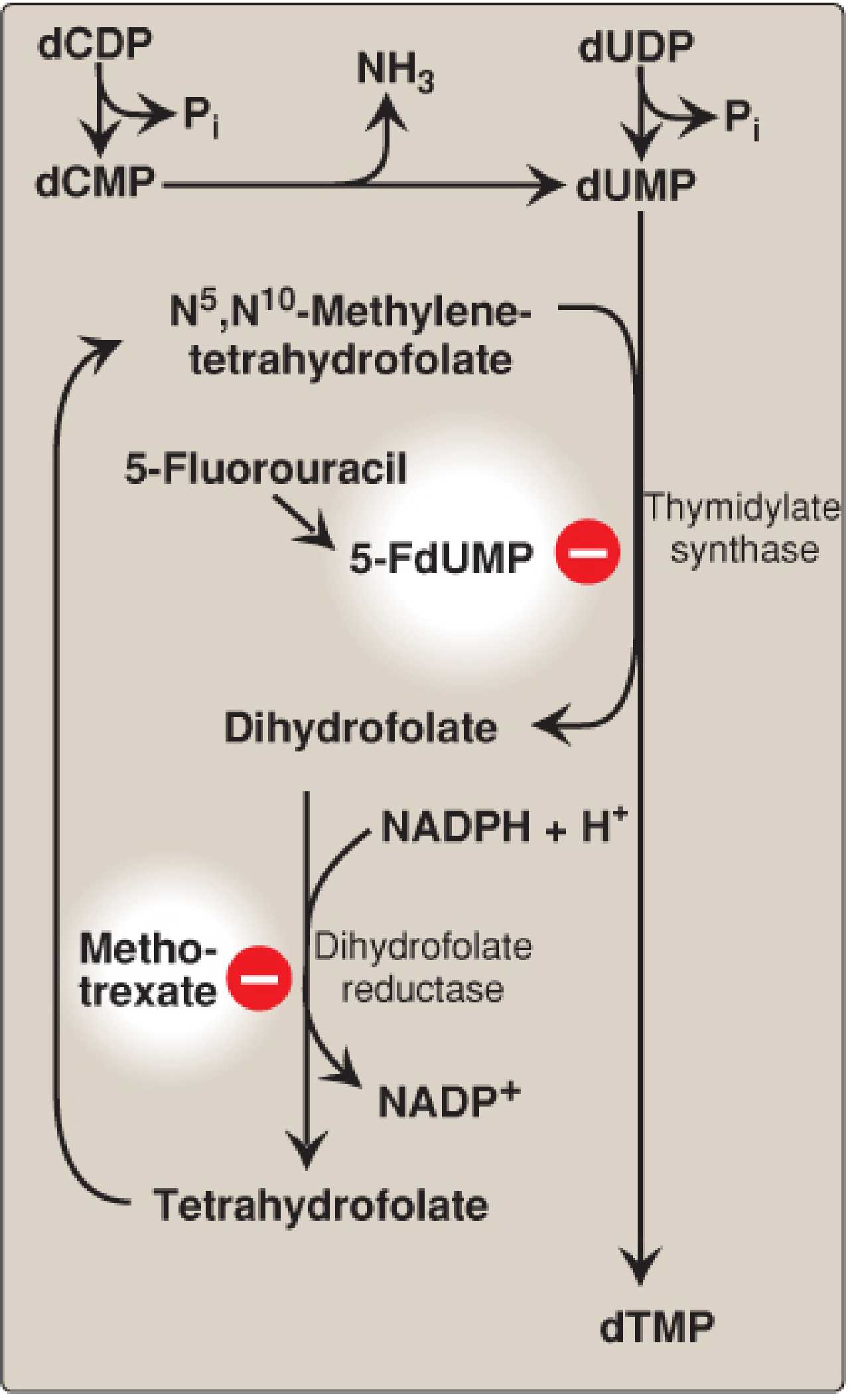
Enzyme: Thymidylate synthase
dUMP + N⁵,N¹⁰-methylene-THF → dTMP + Dihydrofolate (DHF)
This reaction is unique because THF donates both a one-carbon unit (as the methyl group) and two hydrogen atoms from its pteridine ring - resulting in its oxidation to DHF. To regenerate THF, dihydrofolate reductase (DHFR) reduces DHF back using NADPH.
Drug targets at this step:
| Drug | Mechanism | Clinical use |
|---|---|---|
| 5-Fluorouracil (5-FU) | Converted to 5-FdUMP → suicide inhibitor of thymidylate synthase (forms covalent complex with enzyme + methyleneTHF) | Colorectal, breast, head/neck cancer |
| Methotrexate | Folate analog; inhibits DHFR → blocks THF regeneration → depletes dTMP and purine precursors | Cancer, RA, psoriasis |
| Pemetrexed | Inhibits thymidylate synthase AND DHFR AND GARFT (purine synthesis) | Mesothelioma, NSCLC |
Regulation of Pyrimidine Synthesis
The regulated step is CPS II (Step 1):
- Inhibited by UTP (end product feedback inhibition)
- Activated by PRPP (signals need for more nucleotides)
Cell-cycle regulation of CPS II:
- As cells approach S-phase: CPS II becomes more sensitive to PRPP activation and less sensitive to UTP inhibition (phosphorylation by MAP kinase)
- After S-phase: inhibition by UTP is enhanced, PRPP activation reduced (phosphorylation by cAMP-dependent protein kinase)
In prokaryotes, the regulated step is ATCase (Step 2), inhibited by CTP - this is why ATCase is a classical model for allosteric regulation.
Pyrimidine Salvage
Pyrimidines can be recycled but salvage is less clinically significant than purine salvage because the degradation products (β-alanine, β-aminoisobutyrate) are highly water soluble - unlike uric acid from purines.
Two-step salvage:
- Pyrimidine base + ribose 1-phosphate → nucleoside (pyrimidine nucleoside phosphorylase - reverse direction)
- Nucleoside + ATP → nucleotide (nucleoside kinase)
Pyrimidine Degradation
Unlike the purine ring (which stays intact → uric acid), the pyrimidine ring is opened and fully degraded:
- UMP/CMP → cytosine → uracil → β-alanine + NH₃ + CO₂
- TMP → thymine → β-aminoisobutyrate + NH₃ + CO₂
β-alanine and β-aminoisobutyrate are excreted in urine or converted to CO₂, H₂O, and NH₄⁺ (enters urea cycle).
Clinical Diseases
Hereditary Orotic Aciduria
- Deficiency of UMP synthase (one or both activities: orotate phosphoribosyltransferase and/or OMP decarboxylase)
- Cannot convert orotate → OMP → UMP
- Results in: massive orotic acid in urine, failure to thrive, megaloblastic anemia (from pyrimidine deficiency - cells cannot divide; DNA synthesis impaired)
- Treatment: Oral uridine - bypasses the block and is phosphorylated to UMP, from which CTP and dTMP can be produced. Exogenous UMP also provides feedback inhibition of CPS II, reducing orotate accumulation.
OTC Deficiency (urea cycle) causing secondary orotic aciduria
- Carbamoyl phosphate normally used by OTC (ornithine transcarbamylase) in mitochondria accumulates and leaks into the cytosol
- Excess cytosolic carbamoyl phosphate bypasses the regulated CPS II step → excess pyrimidine synthesis → elevated urinary orotate
- Differentiates from hereditary orotic aciduria: in OTC deficiency there is hyperammonemia and the orotic aciduria is secondary
| Feature | Hereditary orotic aciduria | OTC deficiency |
|---|---|---|
| Cause | UMP synthase deficiency | Excess carbamoyl phosphate |
| Orotic acid in urine | ↑↑ | ↑↑ |
| Blood ammonia | Normal | ↑↑ (hyperammonemia) |
| Treatment | Uridine | Protein restriction + nitrogen scavengers |
Summary Pathway
Glutamine + CO₂ + 2ATP ──(CPS II [regulated])──→ Carbamoyl phosphate
│
+ Aspartate (ATCase)
↓
Carbamoylaspartate
│ (Dihydroorotase)
↓
Dihydroorotate
│ (DHODH, mitochondria, FMN)
↓
Orotate
│ + PRPP (orotate PRTase → UMP synthase)
↓
OMP
│ - CO₂ (OMP decarboxylase → UMP synthase)
↓
UMP → UDP → UTP ──(CTP synthetase + Gln)──→ CTP
│
(ribonucleotide reductase)
│
dUDP → dUMP ──(thymidylate synthase + N⁵,N¹⁰-CH₂-THF)──→ dTMP → dTDP → dTTP
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, p. 848-854; Basic Medical Biochemistry - A Clinical Approach - 6e, p. 1425-1432
This is a shared conversation. Sign in to Orris to start your own chat.