Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from Harrison's and Murray & Nadel's textbooks. Let me compile the full answer.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Overview
ARDS is a form of non-cardiogenic pulmonary edema defined by acute hypoxemic respiratory failure with bilateral pulmonary infiltrates that cannot be fully explained by cardiac failure or fluid overload. Unlike cardiogenic edema (driven by elevated hydrostatic pressure), ARDS edema is exudative - the alveolar-capillary barrier becomes abnormally permeable, leaking protein-rich fluid into the airspaces. The result is diffuse alveolar damage (DAD) causing severe ventilation-perfusion mismatch, intrapulmonary shunting, and refractory hypoxemia.
The Berlin criteria classify ARDS by PaO2/FiO2 ratio on at least 5 cmH2O PEEP:
- Mild: 200-300 mmHg
- Moderate: 100-200 mmHg
- Severe: <100 mmHg
Causes: Direct vs. Indirect Lung Injury
| Direct Lung Injury | Indirect (Nonpulmonary) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis (~40-60% of cases) |
| Aspiration of gastric contents | Severe trauma / multiple fractures |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation | Pancreatitis |
| Near-drowning | Drug overdose |
| Reperfusion injury (post-transplant) | Cardiopulmonary bypass |
Over 80% of cases are caused by pneumonia, sepsis, aspiration, trauma, or multiple transfusions. - Harrison's Principles of Internal Medicine 22E, p. 2343
Three Sequential Phases
The natural history unfolds in three overlapping stages:

Time course of ARDS: exudative phase (days 0-7), proliferative phase (days 7-21), and fibrotic phase (>21 days) - Harrison's, p. 2343
Phase 1: Exudative Phase (Days 0-7)
This is the acute injury phase and the cornerstone of ARDS pathogenesis.
Step 1 - Initial alveolar-capillary barrier disruption:
The precipitating insult (direct or via systemic inflammation) damages:
- Capillary endothelial cells - loss of endothelial tight junction integrity
- Type I pneumocytes (ATI cells) - normally covering ~90% of alveolar surface area; their injury is the main driver of barrier failure
The normally tight alveolar barrier becomes highly permeable, allowing protein-rich fluid (with albumin, fibrinogen, coagulation factors) to flood the interstitium and airspaces. - Harrison's, p. 2344
Step 2 - Cytokine storm and neutrophil recruitment:
Resident alveolar macrophages and injured epithelial cells activate Toll-like receptors upon detecting damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs). This drives secretion of pro-inflammatory cytokines:
- IL-1β, IL-6, IL-8, TNF-α - massively elevated in BAL fluid of ARDS patients
- Leukotriene B4 (LTB4) - potent neutrophil chemoattractant
These signals recruit circulating neutrophils (PMNs) into the pulmonary microvasculature and alveoli. - Murray & Nadel's Textbook of Respiratory Medicine, p. 3147
Step 3 - Neutrophil sequestration and transmigration:
One of the earliest findings in ARDS - even before hypoxemia - is transient leukopenia from neutrophil sequestration in the lung microvasculature. Because the average pulmonary capillary diameter is smaller than a neutrophil, they must deform to pass through. Activated neutrophils become "stiff" (due to actin cytoskeletal changes) and cannot negotiate these capillary segments, causing mechanical trapping. Once sequestered, they damage the endothelium and then migrate across into the alveolar space. - Murray & Nadel's, p. 3147
Step 4 - Neutrophil-mediated tissue destruction:
Transmigrated PMNs release a destructive arsenal:
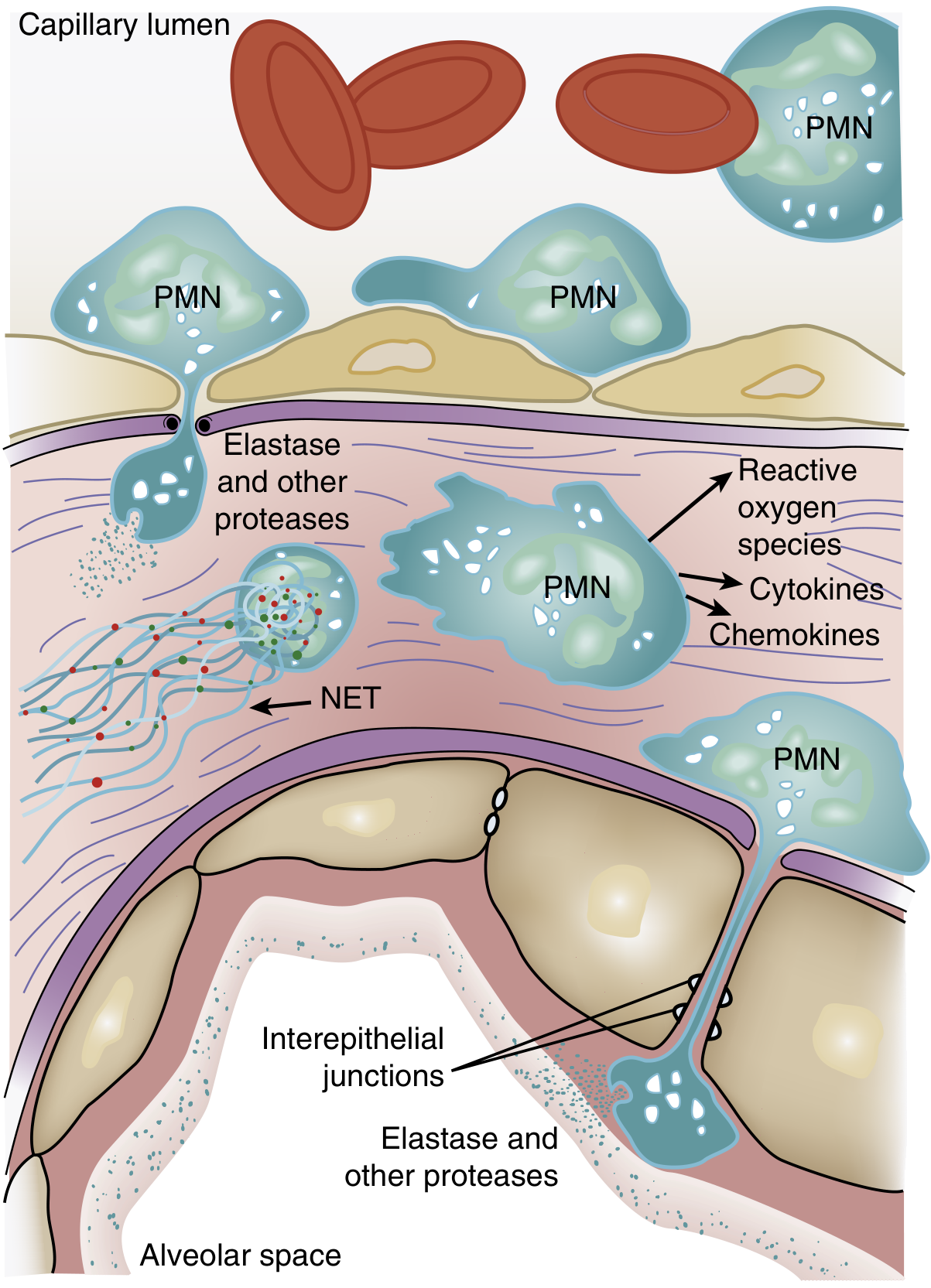
Figure 134.3: Activated PMNs exit the bloodstream, transmigrate across the alveolar-capillary membrane, and release cytokines, proteases, ROS, and NETs - Murray & Nadel's, p. 3147
- Reactive oxygen species (ROS): oxidize lipids, proteins, and DNA in endothelial and epithelial cells
- Neutrophil elastase (NE): degrades epithelial and endothelial cadherins (adherens junction proteins), contributing to barrier breakdown; also degrades growth factors and cytokines, amplifying the inflammatory response
- Matrix metalloproteinases: degrade extracellular matrix
- Neutrophil Extracellular Traps (NETs): web-like structures of DNA, histones, myeloperoxidase, and antimicrobial peptides that trap pathogens but also cause endothelial damage and microthrombus formation. Evidence from mouse models (LPS-induced ARDS, acid aspiration) shows NET formation correlates with severe lung structural destruction, and DNase treatment reduces lung injury.
- Platelet-PMN aggregates: potentiate endothelial injury and contribute to microthrombi
Step 5 - Additional injury pathways:
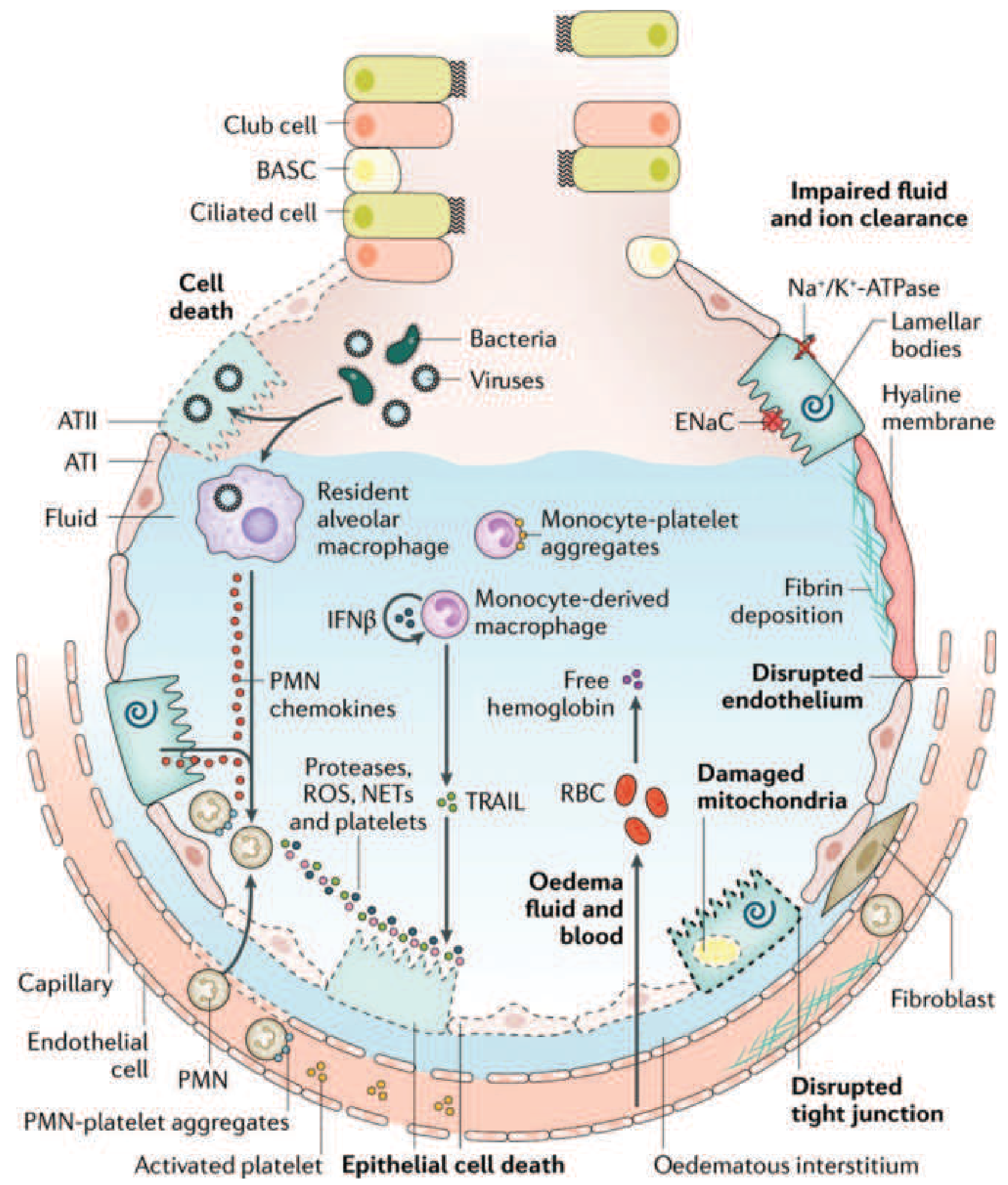
FIGURE 312-3: Comprehensive view of the injured alveolus in ARDS. Multiple converging pathways - neutrophils, monocytes, platelets, free hemoglobin from lysed RBCs, mitochondrial dysfunction, and disrupted tight junctions - all contribute to barrier failure and airspace flooding. Note impaired Na+/K+-ATPase and ENaC function impairs fluid clearance. - Harrison's, p. 2344
- Monocyte-derived macrophages: release IFN-β and TNF-related apoptosis-inducing ligand (TRAIL), causing epithelial apoptosis
- Free hemoglobin from lysed RBCs: exacerbates oxidant-mediated injury
- Fibrin deposition and microvascular occlusion: pulmonary vascular injury causes microthrombi and fibrocellular proliferation, reducing blood flow to ventilated areas
- Surfactant dysfunction: BAL fluid from ARDS patients has markedly abnormal surfactant. Phospholipase A2 (especially relevant in pancreatitis-associated ARDS) degrades surfactant phospholipids. Loss of surfactant function raises alveolar surface tension, promotes alveolar collapse, and further increases vascular permeability. However, surfactant's role is less fundamental in adult ARDS compared to neonatal RDS.
- Impaired alveolar fluid clearance: Hypoxemia and hypercapnia impair Na+/K+-ATPase and epithelial sodium channel (ENaC) function on type II pneumocytes, reducing active resorption of alveolar edema.
Pathophysiologic consequences of the exudative phase:
- Alveoli fill with protein-rich exudate, cellular debris, and hyaline membranes
- Decreased lung compliance (stiff lung)
- Intrapulmonary right-to-left shunting causing refractory hypoxemia
- Increased dead space ventilation (microvascular occlusion prevents blood flow to ventilated units)
- Pulmonary hypertension (hypoxic vasoconstriction, intravascular fibrin, compression by positive-pressure ventilation)
- Hypercapnia from increased dead space
Phase 2: Proliferative Phase (Days 7-21)
Many patients begin to recover during this phase, but others deteriorate.
- Hyaline membranes are reorganized
- Type II pneumocytes (ATII cells) proliferate to replace destroyed ATI cells and attempt to restore the alveolar epithelium (ATII cells are the progenitor cells of the alveolar epithelium)
- Pulmonary fibroblasts migrate in; early fibrosis appears with alveolar-duct and interstitial collagen deposition (elevated N-terminal procollagen peptide III detectable in BAL fluid as early as 24 hours after onset)
- Neutrophil numbers decline; lymphocytes and macrophages predominate
- Persistent dyspnea, tachypnea, and hypoxemia despite some improvement
- Murray & Nadel's, p. 3145; Harrison's, p. 2344
Phase 3: Fibrotic Phase (>21 days, subset of patients)
Not all patients enter this phase, but those who do face high mortality.
- Alveolar and interstitial edema converts to extensive fibrosis with emphysema-like bullae formation
- Acinar architecture is severely disrupted
- Intimal fibroproliferation causes progressive vascular occlusion and pulmonary hypertension
- Decreased lung compliance, increased dead space, risk of pneumothorax
- Lung biopsy evidence of fibrosis at any phase is associated with increased mortality
- Harrison's, p. 2345
Pathophysiology Summary Diagram
| Mechanism | Key Players | Consequence |
|---|---|---|
| Barrier disruption | ATI injury, endothelial tight junction loss | Protein-rich alveolar flood |
| Cytokine storm | TNF-α, IL-1β, IL-6, IL-8, LTB4 | Neutrophil recruitment |
| Neutrophil-mediated destruction | NE, ROS, NETs, metalloproteinases | Epithelial/endothelial death |
| Microvascular injury | Microthrombi, fibrin, hypoxic vasoconstriction | Dead space, pulmonary hypertension |
| Surfactant loss | Phospholipase A2, protein inactivation | Alveolar collapse, shunting |
| Impaired fluid clearance | Na+/K+-ATPase, ENaC dysfunction | Persistent edema |
| Fibrosis (late) | Fibroblast activation, collagen deposition | Irreversible remodeling |
Key Points
- ARDS results from an overwhelming, dysregulated inflammatory response (not just the original insult) damaging the alveolar-capillary unit
- Neutrophils are central to the acute injury; their sequestration is one of the earliest events, even before hypoxemia develops
- The alveolar epithelium (especially ATI cells) plays an underappreciated role - epithelial injury is often more severe than endothelial injury and is a major determinant of outcome
- Three sequential phases (exudative, proliferative, fibrotic) each with distinct histopathology and clinical features
- The "baby lung" concept: CT heterogeneity in ARDS means only a small portion of lung is recruitable - low-tidal-volume ventilation (6 mL/kg ideal body weight) exploits this to reduce ventilator-induced injury, which is the only intervention proven to reduce mortality (31% vs 40% in the ARDS Network trial)
Sources: Murray & Nadel's Textbook of Respiratory Medicine (2-Volume), pp. 3145-3148; Harrison's Principles of Internal Medicine 22E (2025), pp. 2343-2345
This is a shared conversation. Sign in to Orris to start your own chat.