A 52 year old male patient with progressive shrotness of breath and dry coughing since 1 year. Patient in non snoker no history of asthma. On examination velcro crackles hear at the base of ling. Clubbing and cyanosis present. Hrct reveals bilatral opacities with honeycombing. What is diagnosis?
Reading File
Finding Sources
Searching PubMed
"idiopathic pulmonary fibrosis" diagnosis
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
I now have everything needed for a thorough answer. Here it is:
Diagnosis: Idiopathic Pulmonary Fibrosis (IPF)
This is a textbook presentation of IPF. Every clinical, physical, and radiological finding in this case points directly to this diagnosis.
Why Each Finding Points to IPF
| Finding | Significance |
|---|---|
| Age 52, male | IPF classically presents in the 5th-6th decade, more common in men |
| Progressive dyspnoea + dry cough x 1 year | Hallmark presentation - insidious onset |
| Non-smoker, no asthma | Excludes COPD, smoking-related ILD |
| Velcro crackles at lung bases | Pathognomonic inspiratory crackles from fibrotic lung tissue |
| Digital clubbing | Present in ~50% of IPF cases; sign of chronic hypoxia and fibrosis |
| Cyanosis | Reflects significant hypoxaemia from impaired gas exchange |
| HRCT: bilateral opacities + honeycombing | Classic UIP (Usual Interstitial Pneumonia) pattern |
Understanding the HRCT Finding - UIP Pattern
The HRCT showing bilateral opacities with honeycombing represents the Usual Interstitial Pneumonia (UIP) pattern - the radiological hallmark of IPF.
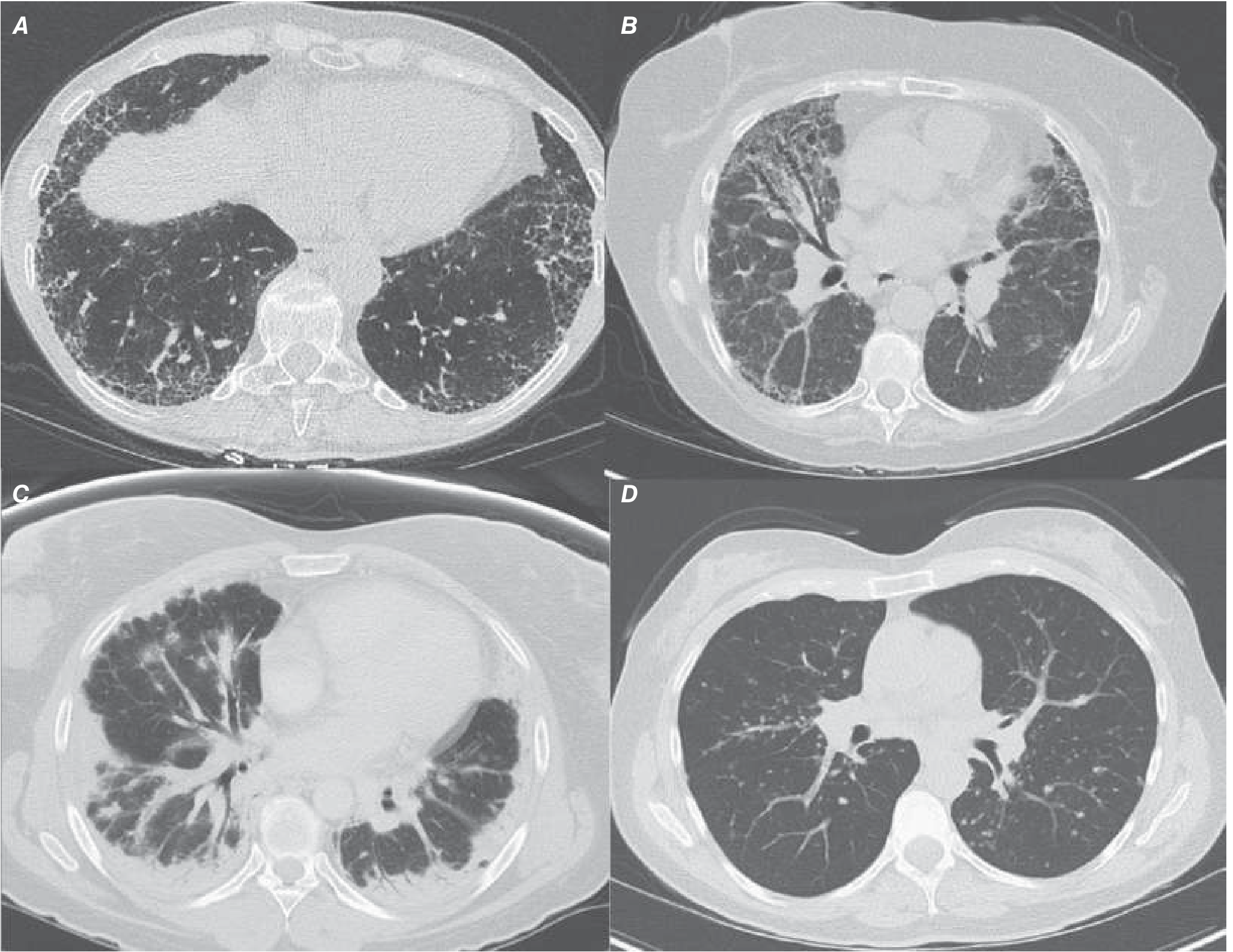
Panel A (top-left) = IPF/UIP: Posterior, basilar, subpleural reticular markings + honeycombing. Panel B = NSIP. Panel C = Cryptogenic Organising Pneumonia. Panel D = Sarcoidosis.
Typical UIP/IPF HRCT features:
- Subpleural, basal-predominant reticulation
- Honeycombing (clustered cystic air spaces, 3-10 mm)
- Traction bronchiectasis
- Bilateral and roughly symmetric distribution
- Posterior > anterior distribution
Ground-glass opacities predominating, bronchovascular changes, upper lobe predominance, or micronodules should raise suspicion for an alternative diagnosis (NSIP, sarcoidosis, hypersensitivity pneumonitis).
Pathophysiology
IPF involves aberrant wound healing with abnormal activation of alveolar epithelial cells, leading to excessive fibroblast proliferation and collagen deposition. The hallmark histological finding is the UIP pattern featuring:
- Temporal and spatial heterogeneity (fibrosis alongside normal lung)
- Fibroblast foci (subepithelial collections of myofibroblasts + collagen)
- Subpleural honeycomb change
- Minimal inflammation
Diagnostic Approach
- HRCT showing typical UIP pattern - sufficient for diagnosis in the right clinical context (no biopsy needed)
- Exclude other causes of ILD: connective tissue disease (CTD), drug toxicity, hypersensitivity pneumonitis, occupational exposure
- PFTs: Restrictive pattern (reduced FVC, TLC, DLCO)
- Surgical lung biopsy (VATS) - if HRCT is atypical or diagnosis uncertain
- BAL - not diagnostic but helps exclude infection, eosinophilic pneumonia
Key Differentials to Exclude
| Condition | How to Distinguish |
|---|---|
| NSIP | More ground-glass opacity, subpleural sparing, affects non-smoking women in 5th decade, responds to steroids |
| Hypersensitivity Pneumonitis | Upper lobe involvement, exposure history, mosaic attenuation |
| CTD-ILD | Positive ANA/anti-Scl70/anti-Jo1, other systemic features |
| Asbestosis | Occupational asbestos exposure history, pleural plaques |
| Sarcoidosis | Upper lobe predominance, hilar lymphadenopathy, bilateral nodules |
Management
Antifibrotic therapy (first-line):
- Nintedanib (tyrosine kinase inhibitor) - slows FVC decline by ~50%
- Pirfenidone - anti-inflammatory and antifibrotic; also slows FVC decline
A recent meta-analysis (Kou et al., 2024) confirmed real-world safety and effectiveness of both agents, reinforcing their use as standard of care.
Supportive care:
- Supplemental oxygen (improves exercise tolerance, reduces pulmonary hypertension risk)
- Pulmonary rehabilitation
- Treat comorbidities (GERD, pulmonary hypertension, OSA)
- Avoid immunosuppression - shown to increase morbidity and mortality in IPF
Lung transplantation:
- Only curative option
- Reserved for eligible patients with progressive disease
- Can improve survival and quality of life
Prognosis
IPF carries a poor prognosis. Median survival is 3-5 years from diagnosis, with approximately 50% 3-to-5-year survival. The disease is variably progressive - some patients decline slowly, others experience acute exacerbations with rapid deterioration.
Sources: Harrison's Principles of Internal Medicine, 22e (2025), p. 2305-2306; Murray & Nadel's Textbook of Respiratory Medicine
This is a shared conversation. Sign in to Orris to start your own chat.