apoptosis
Reading File
Finding Sources
Searching PubMed
apoptosis mechanisms pathways
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from Robbins Pathology and Sleisenger's GI textbook, plus two excellent diagrams. Here is the full answer:
Apoptosis
Apoptosis is a form of programmed cell death in which cells activate intrinsic enzymatic machinery to destroy their own nuclear DNA, proteins, and cytoskeletal components. Unlike necrosis, the plasma membrane remains intact throughout the process; the dying cell is packaged into membrane-bound fragments called apoptotic bodies that are rapidly phagocytosed by macrophages - with no leakage of cell contents and therefore no inflammatory response.
Morphological Features
- Chromatin compaction and condensation (pyknosis)
- Nuclear fragmentation (karyorrhexis)
- Cytoplasmic condensation
- Plasma membrane blebbing and budding off of apoptotic bodies
- Rapid phagocytosis - no inflammation
This is the key distinction from necrosis, where the cell swells, membrane ruptures, and contents spill out, triggering inflammation.
Two Major Pathways
Both pathways converge on activation of caspases - cysteine proteases that cleave after aspartate residues - ultimately activating executioner caspases 3 and 7.
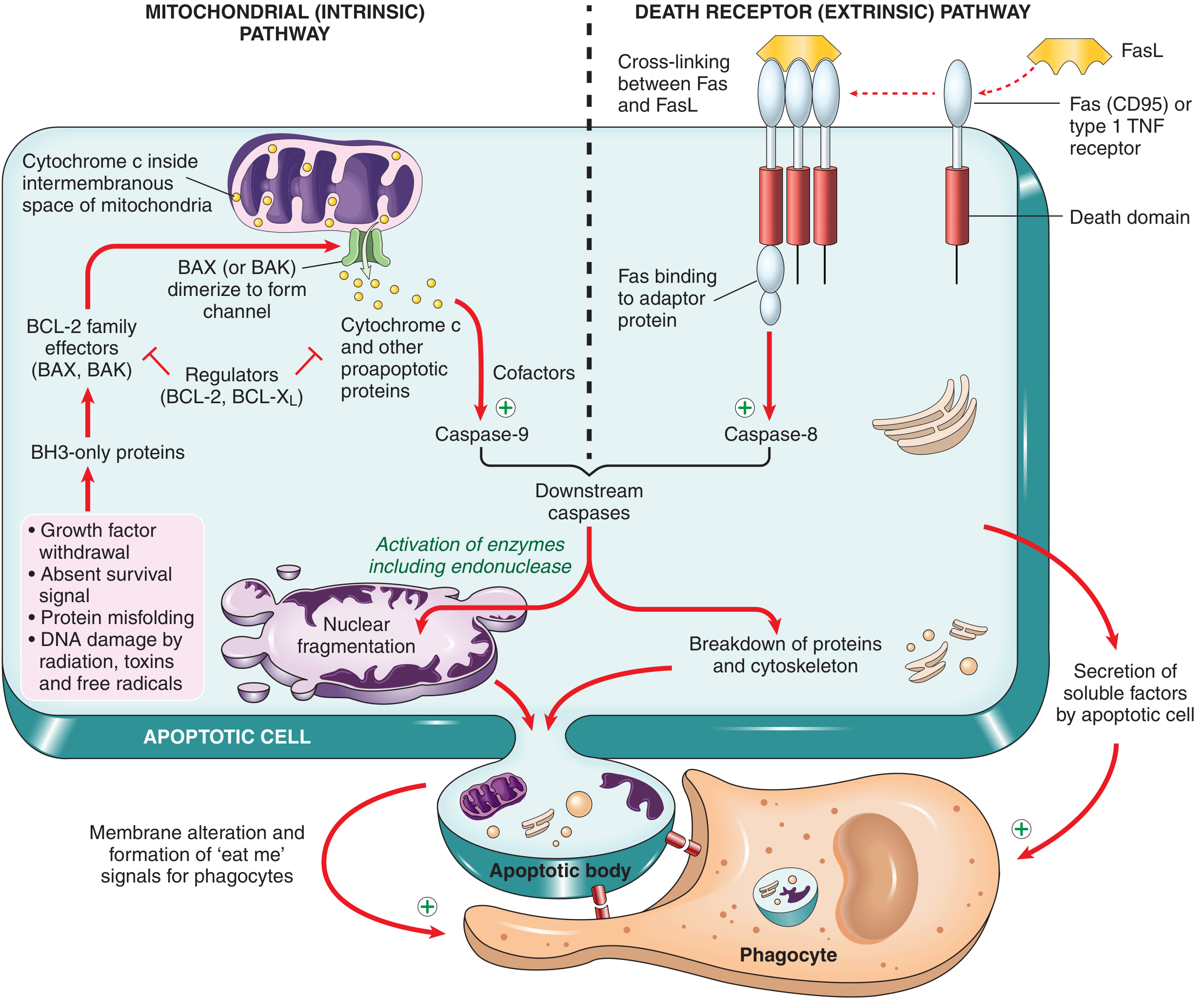
1. Mitochondrial (Intrinsic) Pathway
This is the dominant pathway in most physiologic and pathologic apoptosis.
Triggers:
- Growth factor or survival signal withdrawal
- DNA damage (radiation, toxins, free radicals)
- Protein misfolding / ER stress
- Hypoxia, nutrient deprivation
Mechanism:
- Stress signals activate BH3-only proteins (sensors of the BCL-2 family, e.g., BID, BAD, BIM, PUMA, NOXA)
- BH3-only proteins shift the balance toward proapoptotic effectors BAX and BAK, which dimerize and insert into the outer mitochondrial membrane, forming channels
- Mitochondrial outer membrane permeabilization (MOMP) allows cytochrome c to leak into the cytosol
- Cytochrome c + APAF-1 + procaspase-9 form the apoptosome
- The apoptosome activates caspase-9, which cleaves and activates executioner caspases 3 and 7
Anti-apoptotic checkpoint: BCL-2 and BCL-XL (induced by growth factors) normally hold BAX/BAK in check and maintain mitochondrial membrane integrity. Cancer cells often overexpress BCL-2 (e.g., follicular lymphoma - t(14;18) translocation).
2. Death Receptor (Extrinsic) Pathway
Triggered by ligation of cell surface death receptors of the TNF receptor superfamily, including:
- Fas (CD95) bound by FasL
- TNFR1 bound by TNF
Mechanism:
- Receptor cross-linking induces clustering of death domains in the cytoplasmic tails
- Adaptor proteins (e.g., FADD) are recruited, forming the DISC (Death-Inducing Signaling Complex)
- DISC activates caspase-8 (initiator caspase)
- Caspase-8 directly activates executioner caspases 3 and 7
Note: Caspase-8 can also cleave BID to generate tBID, which amplifies the signal through the mitochondrial pathway - this is the crossover point between the two pathways.
Downstream Execution
Once executioner caspases (3 and 7) are active, they cleave:
- Endonucleases - activate DNase that cleaves DNA at internucleosomal linker regions (produces the classic "DNA ladder" on gel electrophoresis)
- Nuclear lamins - causes nuclear fragmentation
- Cytoskeletal proteins - causes cell shape changes and blebbing
- IAP inhibitors - amplifies caspase activity
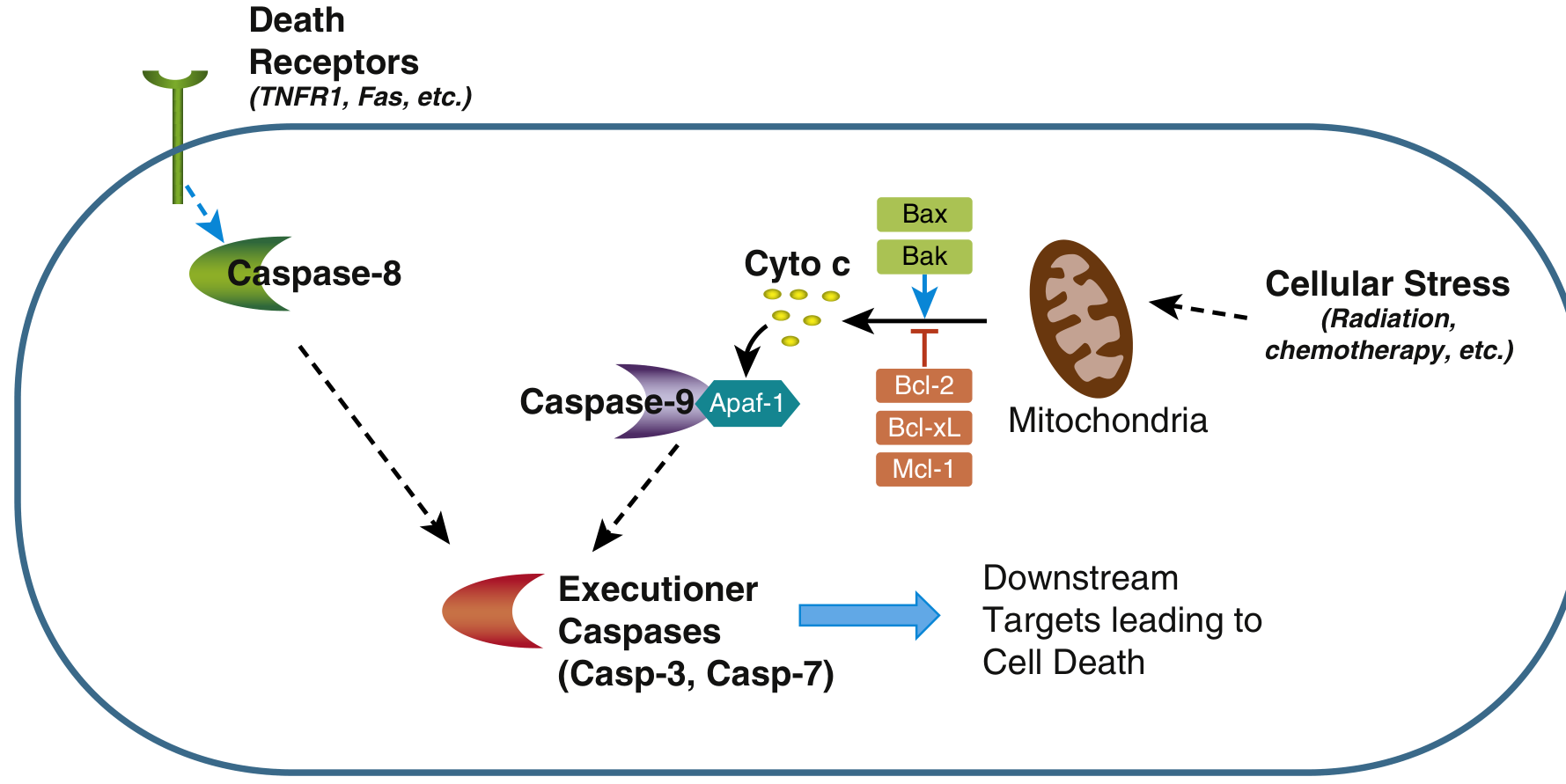
BCL-2 Family - The Master Regulators
| Subfamily | Members | Function |
|---|---|---|
| Anti-apoptotic | BCL-2, BCL-X | Prevent MOMP; inhibit BAX/BAK |
| Proapoptotic effectors | BAX, BAK | Form pores in mitochondrial membrane |
| BH3-only sensors | BIM, BID, BAD, PUMA, NOXA | Activate BAX/BAK; neutralize BCL-2 |
Physiologic Roles of Apoptosis
| Situation | Mechanism |
|---|---|
| Embryonic development (tissue patterning) | Growth factor withdrawal |
| Turnover of intestinal epithelium, lymphocytes | Loss of survival signals |
| Involution of hormone-dependent tissues (endometrium) | Reduced hormone levels |
| Decline of immune cells after an immune response | Loss of survival signals |
| Deletion of self-reactive lymphocytes (central tolerance) | Fas/FasL and mitochondrial pathways |
Pathologic Apoptosis
- Increased apoptosis: neurodegenerative diseases (Alzheimer's, Parkinson's - loss of neurons), viral infections (cytotoxic T lymphocytes kill infected cells via Fas/perforin-granzyme), ischemia-reperfusion injury
- Deficient apoptosis: cancer (BCL-2 overexpression, p53 mutations impair BH3-only protein induction), autoimmune diseases (failure to eliminate self-reactive lymphocytes - mutations in Fas cause autoimmune lymphoproliferative syndrome, ALPS)
Caspase Classification
| Type | Members | Role |
|---|---|---|
| Initiator caspases | Caspase-8, -9 | Activated by DISC or apoptosome; activate executioners |
| Executioner caspases | Caspase-3, -7 | Cleave downstream substrates causing cell death |
Caspases are synthesized as inactive zymogens (procaspases) and activated by proteolytic cleavage - either by another caspase or by focal accumulation of procaspase monomers that self-activate.
Clinical Relevance
- Venetoclax - BCL-2 inhibitor; used in CLL and AML (restores apoptosis in cancer cells)
- TRAIL-based therapies - activate death receptors selectively in tumor cells
- p53 is the chief activator of proapoptotic BH3-only proteins (e.g., PUMA) in response to DNA damage; loss of p53 (the most common mutation in cancer) disables this checkpoint
- Granzyme B from cytotoxic T cells enters target cells via perforin pores and directly activates caspases
Sources:
- Robbins & Kumar Basic Pathology, p. 24-26
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease, p. 23
Recent reviews: Mustafa et al. (2024) provide a comprehensive update on apoptosis signaling in Cells, and Moyer et al. (2025) review apoptosis in cancer biology in Annual Review of Pathology - both consistent with the mechanisms described above.
This is a shared conversation. Sign in to Orris to start your own chat.