Pheochromocytoma
Reading File
Reading File
Finding Sources
Searching PubMed
pheochromocytoma diagnosis management
Running Command
Finding Sources
Reading File
Running Command
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Searching Images
pheochromocytoma adrenal tumor catecholamine hypertension
I now have comprehensive material from the textbook and images. Let me compile the full response on pheochromocytoma.
Pheochromocytoma
A pheochromocytoma is a catecholamine-secreting neuroendocrine tumor arising from chromaffin cells of the adrenal medulla. Extra-adrenal tumors arising from sympathetic or parasympathetic paraganglia are termed paragangliomas and share a similar biology and management approach.
Gross and Histologic Pathology
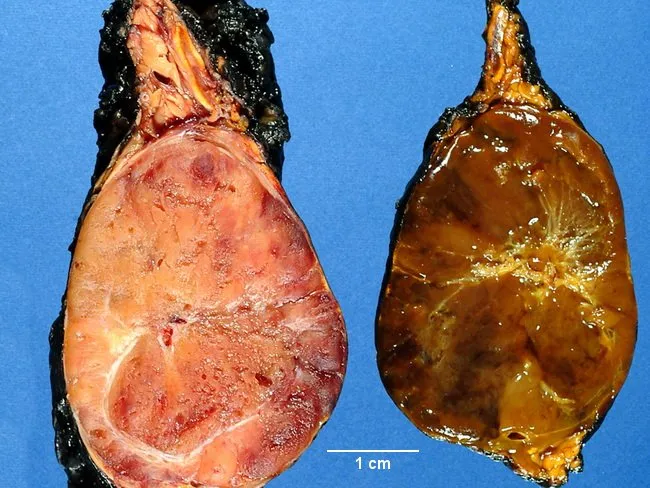
- Gross: Pink-tan to yellow, well-circumscribed mass; may show hemorrhage and necrosis. Positive chromaffin reaction (darkening on potassium dichromate exposure) confirms catecholamine content.
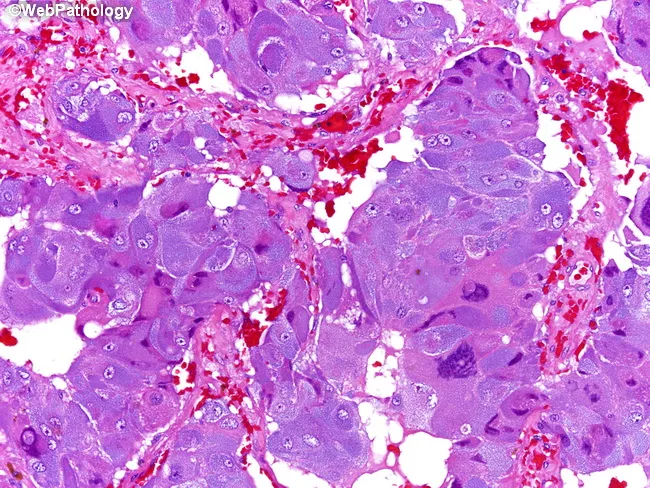
- Histology: Characteristic zellballen (cell-ball nesting) of polygonal chromaffin cells with granular eosinophilic cytoplasm, round nuclei with "salt-and-pepper" chromatin, and a rich capillary stroma. Sustentacular cells at nest periphery are S-100 positive on IHC.
- IHC: Chromogranin A, synaptophysin, and NSE positive.
Epidemiology
- Incidence: ~1–2 per 100,000 per year; found in ~0.1–0.6% of hypertensive patients.
- The old "10% rule" (10% bilateral, 10% malignant, 10% extra-adrenal, 10% familial) is now outdated — genetic discoveries have substantially revised these proportions upward.
- In pediatric patients: more frequently bilateral, less malignant, and more commonly associated with hereditary syndromes (MEN1, MEN2A, MEN2B).
- Extra-adrenal tumors (paragangliomas) most commonly occur in the organ of Zuckerkandl (aortic bifurcation), and lack PNMT, so they secrete only norepinephrine (not epinephrine).
Clinical Features
Classic Triad
Headache + Diaphoresis + Palpitations in a hypertensive patient is highly suggestive.
Hypertension Pattern
- Sustained hypertension (most common, ~50%)
- Paroxysmal hypertensive crises (episodic)
- Rarely, normotension between episodes
Additional Features
| System | Manifestations |
|---|---|
| Cardiovascular | Tachycardia, arrhythmias, cardiomyopathy, hypertensive emergency |
| Metabolic | Hyperglycemia, weight loss |
| GI | Nausea, abdominal pain, constipation |
| Constitutional | Pallor (vasoconstriction), anxiety, tremor |
| Ophthalmologic | Hypertensive retinopathy |
Paroxysmal attacks may be triggered by: tumor manipulation, micturition (bladder paraganglioma), exercise, anesthesia induction, opioids, histamine, glucagon, or tyramine-containing foods if on MAOIs.
Biochemical Diagnosis
Catecholamine excess is confirmed by measuring catecholamines and their O-methylated metabolites (metanephrines):
| Test | Sensitivity | Specificity | Notes |
|---|---|---|---|
| Plasma free metanephrines | ~99% | ~85–89% | Best initial screen — excellent for ruling OUT |
| 24-hr urine metanephrines + catecholamines | ~88% | High | Confirmatory; cutoffs set at ~2× upper reference range |
| Chromogranin A | Moderate | Moderate | Marker of neuroendocrine tumors |
Key point: Plasma free metanephrines have such high sensitivity that a negative result effectively excludes pheochromocytoma. However, false-positive rates are high (false positives may outnumber true positives by 30:1 in hypertensive screening populations), so confirmatory 24-hr urine testing is essential when plasma screening is positive.
Confounders of Catecholamine Testing
- Sympathomimetics (cold remedies)
- Phenoxybenzamine (itself raises catecholamines)
- Acetaminophen (interferes with plasma free metanephrine assay)
- Tricyclic antidepressants, other psychotropics
- Acute pain, critical illness, emotional stress
Imaging & Localization
Once biochemical diagnosis is established, imaging proceeds:
- CT abdomen/pelvis (first-line): High sensitivity (~90–100%) for adrenal lesions; ~4–5 cm mean size; may show heterogeneous enhancement, central necrosis.
- MRI: Characteristic T2 hyperintensity ("light bulb" appearance); preferred in pregnancy, children, or when avoiding radiation.
- Functional imaging for extra-adrenal, multifocal, or metastatic disease:
- ¹²³I-MIBG scintigraphy (norepinephrine analogue; uptake by chromaffin cells)
- ⁶⁸Ga-DOTATATE PET (increasingly preferred; higher sensitivity than MIBG)
- ¹⁸F-FDG PET — for SDHB-mutated or malignant disease
MIBG is contraindicated in pregnancy (radiation exposure).
Perioperative Management
Alpha-Blockade (MANDATORY before surgery)
Preoperative α-adrenergic blockade is essential to prevent intraoperative hypertensive crisis:
| Agent | Mechanism | Notes |
|---|---|---|
| Phenoxybenzamine | Non-selective, irreversible α-blocker | Gold standard; causes nasal stuffiness, orthostatic hypotension |
| Doxazosin/Prazosin | Selective α₁-blocker | Better tolerated; commonly used |
| Calcium channel blockers | Adjunct | Added if blood pressure uncontrolled on α-blockade alone |
Beta-Blockade
- Added only after α-blockade is established (never first — unopposed α-stimulation worsens hypertension)
- Controls reflex tachycardia
High-Salt / High-Fluid Diet
- Given during α-blockade to expand intravascular volume (combats the vasoconstriction-induced volume contraction)
Duration
Typically 1–2 weeks preoperatively; target BP <130/80 with mild orthostatic hypotension acceptable.
Surgical Management
- Laparoscopic adrenalectomy is the standard of care for most pheochromocytomas (≤6–8 cm).
- Posterior retroperitoneoscopic adrenalectomy increasingly used — shorter operative time, less bowel manipulation.
- Open approach for large (>8–10 cm), locally invasive, or suspected malignant tumors.
- Intraoperatively: careful anesthetic management; IV phentolamine or nitroprusside for hypertensive crises; magnesium sulfate as adjunct.
- Following tumor removal: expect hypotension from catecholamine withdrawal — IV fluid resuscitation, vasopressors if needed.
Cortical-Sparing (Partial) Adrenalectomy
- Preferred in hereditary/bilateral disease (MEN2, VHL) to preserve adrenal cortex and avoid permanent Addison's disease.
- A 2025 systematic review and meta-analysis (PMID 40214691) confirms favorable surgical outcomes for partial adrenalectomy for pheochromocytoma.
Molecular Genetics
At least one-third of pheochromocytoma patients carry a germline mutation — genetic testing is indicated for ALL patients.
| Syndrome | Gene | Additional Features |
|---|---|---|
| MEN2A / MEN2B | RET | Medullary thyroid cancer ± parathyroid hyperplasia; bilateral pheo in 40–50% |
| Von Hippel-Lindau | VHL | Hemangioblastomas, clear cell RCC, pancreatic cysts; pheo in 10–20% |
| Neurofibromatosis type 1 | NF1 | Café-au-lait spots, neurofibromas, Lisch nodules; pheo in 1–5% |
| Familial paraganglioma | SDHA/B/C/D, SDHAF2, TMEM127 | GI stromal tumors, renal cell carcinoma |
| HIF2A/EPAS1 mutation | HIF2A | Familial polycythemia, somatostatinomas |
| MAX mutation | MAX | Unknown additional features |
| HLRCC | FH | Leiomyomas, renal cell carcinoma |
SDHB mutation specifically confers high risk of extra-adrenal location and malignancy — these patients need aggressive surveillance.
Familial cases: earlier age at presentation, higher rate of bilaterality and multifocality.
Metastatic (Malignant) Pheochromocytoma
- Malignancy is defined solely by the presence of metastases (no reliable histologic criteria distinguish benign from malignant).
- Most common metastatic sites: lymph nodes, bone, liver, lung.
- Higher risk: extra-adrenal location, large size (>5 cm), SDHB mutation, dopamine-secreting tumors.
- MIBG Score / PASS score (Pheochromocytoma of the Adrenal gland Scaled Score) attempts risk stratification.
Management Options for Metastatic Disease
| Modality | Notes |
|---|---|
| ¹³¹I-MIBG therapy | First-line for MIBG-avid disease |
| Sunitinib / CVD chemotherapy | Cyclophosphamide-vincristine-dacarbazine (CVD); moderate response rates |
| Peptide receptor radionuclide therapy (PRRT) | ¹⁷⁷Lu-DOTATATE for somatostatin receptor-positive tumors |
| Temozolomide | Particularly in SDHB-mutated tumors |
| Debulking surgery | Symptom control |
Special Situations
Pregnancy
- Potentially fatal for mother and fetus if unrecognized (27-fold increase in maternal/fetal complications).
- α-Blockade is mandatory; MIBG is contraindicated.
- If diagnosed < 24 weeks: laparoscopic adrenalectomy in the 2nd trimester may be considered.
- If diagnosed ≥ 24–28 weeks: defer surgery; deliver by cesarean section, then perform adrenalectomy.
Incidental Adrenal Mass (Incidentaloma)
- Screen all adrenal incidentalomas with plasma free metanephrines to exclude pheochromocytoma before any biopsy or procedure (biopsy of an unsuspected pheochromocytoma can precipitate fatal hypertensive crisis).
MEN2 – Operative Sequencing
- If pheo identified alongside medullary thyroid cancer (MTC): adrenalectomy FIRST to normalize catecholamines, then thyroidectomy.
Postoperative Follow-up
- Biochemical testing (plasma/urine metanephrines) at 2–4 weeks postop to confirm cure.
- Annual surveillance for 10 years (longer for hereditary/SDHB cases) given risk of recurrence.
- Genetic counseling and cascade family testing when germline mutation identified.
Sources: Sabiston Textbook of Surgery (Diseases of the Adrenal Medulla); Nelson Textbook of Pediatrics 2024 (Endocrine System); Araujo-Castro et al., Rev Endocr Metab Disord 2025 — Surgical outcomes of partial adrenalectomy for pheochromocytoma, PMID 40214691.
Pheochromocytoma from Nelson text book
Running Command
Running Command
Running Command
Running Command
Running Command
Running Command
Running Command
I now have the complete Nelson 2024 Chapter 621 on Pheochromocytoma (pages 124–125) plus the genetics table (page 118). Here is the full presentation directly from the textbook:
Pheochromocytoma
Nelson Textbook of Pediatrics, 2024 — Chapter 621
Author: Perrin C. White
Definition
Pheochromocytomas are catecholamine-secreting tumors arising from chromaffin cells. The most common site of origin (~90%) is the adrenal medulla; however, tumors may develop anywhere along the abdominal sympathetic chain — most often near the aorta at the level of the inferior mesenteric artery or at its bifurcation. They also appear in the periadrenal area, urinary bladder or ureteral walls, thoracic cavity, and cervical region.
Epidemiology
- 10% of all pheochromocytomas occur in children, presenting most frequently between 6 and 14 years of age.
- Tumors vary from 1 to 10 cm in diameter; found more often on the right side than the left.
- In children, the pattern is different from adults:
- >20% have bilateral adrenal tumors.
- 30–40% have tumors in both adrenal and extraadrenal areas, or only in an extraadrenal area.
- Most pheochromocytomas are associated with germline pathogenic variants.
Genetics & Associated Syndromes
Most pheochromocytomas are hereditary. Key associations:
| Syndrome | Gene | Additional Features |
|---|---|---|
| Von Hippel-Lindau (1 in 36,000) | VHL (chr 3p25-26) | Retinal & CNS hemangioblastomas, renal clear cell carcinoma; pheo in some kindreds only |
| MEN2A / MEN2B | RET (chr 10q11.2) | Medullary thyroid carcinoma, parathyroid tumors; ~50% develop pheo; codon 634 variants = highest risk |
| Neurofibromatosis type 1 | NF1 (chr 17q11.2) | Café-au-lait spots, neurofibromas, Lisch nodules; occasional pheo |
| Familial paraganglioma | SDHB, SDHD, SDHC, SDHA, SDHAF2, MAX, TMEM127 | Paragangliomas, often head/neck; ~50% of SDHB-mutant tumors are malignant |
| Carney-Stratakis dyad | SDH genes | Pheo/paraganglioma + GI stromal tumors |
| Carney-Stratakis triad | SDH genes | Pheo/paraganglioma + GISTs + pulmonary chondromas |
| 3P Association (3Pas) | SDH genes | Pheo/paraganglioma + pituitary adenomas |
The VHL and SDH gene products both participate in the pseudohypoxia signaling pathway (decreased NAD⁺/NADH ratio), representing a common pathogenetic pathway.
Clinical Manifestations
Tumors detected by surveillance in known mutation carriers may be asymptomatic. When symptomatic, hypertension is the cardinal feature — resulting from excessive secretion of metanephrines, epinephrine, and norepinephrine.
All patients have hypertension at some point.
Pediatric vs. Adult Pattern
- In children: hypertension is more often sustained rather than paroxysmal (unlike adults).
- Paroxysmal attacks start infrequently, become more frequent, and eventually give way to a continuous hypertensive state.
- Between attacks: patient may be symptom-free.
Symptoms During Attacks
- Headache, palpitations, abdominal pain, dizziness
- Pallor, vomiting, sweating
- Seizures and hypertensive encephalopathy
- Severe cases: precordial pain radiating to arms, pulmonary edema, cardiac and hepatic enlargement
Other Features
- Good appetite but no weight gain (hypermetabolic state); severe cachexia may develop
- Polyuria and polydipsia — can mimic diabetes insipidus
- Growth failure (striking in children)
- BP: systolic 180–260 mmHg / diastolic 120–210 mmHg
- Ophthalmoscopy: papilledema, hemorrhages, exudate, arterial constriction
- Symptoms worsened by exercise or stimulants (e.g., pseudoephedrine in OTC medications)
Laboratory Findings
| Finding | Detail |
|---|---|
| Urine | Proteinuria, casts, occasionally glucose; gross hematuria → bladder wall tumor |
| Polycythemia | Occasionally observed |
| Catecholamine secretion | Children predominantly excrete norepinephrine (unlike adults who have both ↑NE and ↑Epi) |
| VMA | ↑ urinary vanillylmandelic acid (3-methoxy-4-hydroxymandelic acid), but no longer routinely measured due to food interference (vanilla, fruits) |
Preferred Biochemical Tests
Consensus: measure plasma free metanephrines AND urinary fractionated metanephrines.
Important pre-test instructions:
- Abstain from caffeinated drinks
- Avoid acetaminophen (interferes with plasma normetanephrine immunoassay)
- Draw blood from indwelling IV catheter to avoid acute stress of venipuncture
Imaging & Localization
- CT or MRI — most adrenal-area tumors readily localized (Fig. 621.1: bilateral pheo in VHL showing T2-hyperintense masses)
- ¹²³I or ¹³¹I-MIBG — taken up by chromaffin tissue anywhere in the body; useful for small or extraadrenal tumors
- PET-CT — ¹⁸F-FDG or ⁶⁸Ga-DOTATATE (somatostatin receptor ligand) — highly sensitive and increasingly preferred for difficult-to-localize tumors (Fig. 621.2: paraganglioma at Zuckerkandl organ showing T2 "light bulb" sign on MRI and MIBG uptake on SPECT/CT)
- Venous catheterization with catecholamine sampling — now only rarely necessary
MRI findings: characteristic T2-hyperintensity ("light bulb" sign) in pheochromocytomas and paragangliomas; heterogeneous contrast enhancement.
Differential Diagnosis
Causes of hypertension in children to consider:
- Renal or renovascular disease
- Coarctation of the aorta
- Hyperthyroidism
- Cushing syndrome
- Enzyme deficiencies: 11β-hydroxylase, 17α-hydroxylase, 11β-HSD type 2
- Primary aldosteronism; adrenocortical tumors
- Porphyria or familial dysautonomia (for paroxysmal hypertension)
- Essential hypertension (rare in children)
- Neuroblastoma, ganglioneuroblastoma, ganglioneuroma: also produce catecholamines, but dopamine and HVA levels are usually higher in neuroblastoma, while most catecholamine levels are higher in pheochromocytoma
Treatment
Surgical Removal (Definitive)
These tumors must be removed surgically; careful preoperative, intraoperative, and postoperative management is essential.
Manipulation and excision cause marked ↑ catecholamine release → ↑ BP and ↑ HR.
Preoperative Medical Management
| Drug Class | Agent | Notes |
|---|---|---|
| Non-selective α-blocker | Phenoxybenzamine | Most widely used; may not be covered by insurance |
| Selective α₁-blocker | Doxazosin | Alternative to phenoxybenzamine |
| Calcium channel blocker | Amlodipine | Also used as alternative/adjunct |
| β-blockade | Added after α-blockade | Controls reflex tachycardia |
Volume expansion with appropriate fluids before and during surgery is critical to avoid precipitous BP drop intraoperatively or within 48 hours postoperatively.
Surgical Approach
- Because tumors are often multiple in children, a thorough transabdominal exploration of all usual sites offers the best opportunity to find all tumors.
- Cortical-sparing (adrenal-sparing) surgery has been advocated for bilateral/recurrent disease to reduce risk of Addison disease — however, this increases risk of tumor recurrence.
Malignancy
- Histology alone is unreliable for predicting malignancy.
- True malignancy is defined by metastatic disease or local invasiveness precluding complete resection.
- ~10% of adrenal pheochromocytomas are malignant overall; rare in childhood.
- Pediatric malignant pheochromocytomas: more frequent at extraadrenal sites and often associated with SDHB pathogenic variants.
Follow-up
- Prolonged follow-up is mandatory — particularly in patients with germline mutations, as functioning tumors at other sites may appear many years after the initial operation.
- Examine relatives of affected patients — unsuspected, asymptomatic tumors may be found.
Source: Nelson Textbook of Pediatrics, 2024, Chapter 621 — Pheochromocytoma (Perrin C. White), Part XXIV: The Endocrine System, pp. 3479–3480.
This is a shared conversation. Sign in to Orris to start your own chat.