classify cutaneous vasculitis .Discuss Henoch Scholein purpura in 10marks format for 3rd yr dermatology post graduate theory examination
Reading File
Finding Sources
Finding Sources
Searching PubMed
IgA vasculitis Henoch-Schonlein purpura
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive data from all major dermatology textbooks. Let me compile the complete, well-structured 10-mark answer:
Classification of Cutaneous Vasculitis & Henoch-Schönlein Purpura
PART I: CLASSIFICATION OF CUTANEOUS VASCULITIS
The cornerstone of classification is vessel size, as established by the 2012 revised International Chapel Hill Consensus Conference (CHCC) Nomenclature, with a 2018 dermatologic addendum. Clinical morphology of skin lesions directly correlates with the caliber of the affected vessel.
A. Based on Vessel Size (Table — Chapel Hill / Dermatology 5e)
| Vessel Size | Vasculitis Type | Subtypes / Causes | Cutaneous Morphology |
|---|---|---|---|
| Small (arterioles, capillaries, post-capillary venules; superficial/mid-dermis) | Small Vessel Vasculitis (SVV) | • Cutaneous Small Vessel Vasculitis (CSVV) — idiopathic | Palpable purpura (most common), petechiae, macular purpura, urticarial plaques, vesicles, pustules, targetoid papules, round ulcers |
| • IgA vasculitis (Henoch-Schönlein purpura) | |||
| • Acute hemorrhagic edema of infancy | |||
| • Urticarial vasculitis | |||
| • Erythema elevatum diutinum | |||
| Secondary: Drugs, Infections (septic vasculitis), Autoimmune-CTD, Hematologic malignancies | |||
| Small + Medium ("Mixed") | ANCA-associated vasculitis | • Granulomatosis with polyangiitis (GPA/Wegener's) | Petechiae, palpable purpura, livedo racemosa, retiform purpura, ulcers, subcutaneous nodules, digital necrosis |
| • Microscopic polyangiitis (MPA) | |||
| • Eosinophilic GPA (Churg-Strauss) | |||
| Cryoglobulinemic vasculitis (types II & III) | Secondary to HCV, AI-CTD, lymphoproliferative disorders | ||
| Medium (small arteries/veins; deep dermis/subcutis) | Polyarteritis nodosa (PAN) | • Classic (systemic) PAN | Livedo racemosa, retiform purpura, ulcers, subcutaneous nodules, digital necrosis |
| • Cutaneous PAN (cutaneous arteritis) | |||
| Secondary: Infections, AI-CTD | |||
| Large (aorta and named arteries) | Temporal (Giant Cell) Arteritis | — | Tender scalp nodules, frontotemporal ulceration/gangrene, alopecia |
| Takayasu Arteritis | — | Rare cutaneous involvement |
Cutaneous involvement occurs almost exclusively with small and medium-vessel vasculitis. Behçet disease may involve vessels of all sizes. — Dermatology 2-Volume Set 5e (Bolt/Griffiths); Fitzpatrick's Dermatology Vol. 1 & 2
B. Histopathologic Terms
| Term | Meaning |
|---|---|
| Leukocytoclastic vasculitis (LCV) | Histologic pattern — neutrophilic inflammation of post-capillary venules with karyorrhexis ("nuclear dust"), fibrinoid necrosis; not synonymous with CSVV |
| CSVV (Cutaneous Small Vessel Vasculitis) | Clinical-pathologic entity: single-organ, skin-limited SVV |
| Pauci-immune vasculitis | ANCA-associated; vessel damage mediated by neutrophils, NOT by immune complex deposition |
C. By Etiology (Etiologic Classification)
- Idiopathic (~50% of CSVV cases)
- Drug-induced (~20%): Almost any drug; PTU, hydralazine (ANCA-associated)
- Infection-associated: Group A β-hemolytic Streptococcus, Mycoplasma, HCV, H. pylori
- Autoimmune-CTD associated: SLE (10–36%), Sjögren syndrome (10%), RA
- Malignancy-associated: Solid tumors, lymphoproliferative neoplasms
- Systemic vasculitis with cutaneous involvement: GPA, MPA, PAN, IgA vasculitis
PART II: HENOCH-SCHÖNLEIN PURPURA (IgA VASCULITIS) — 10 MARKS FORMAT
1. Definition & Nomenclature
IgA Vasculitis (IgAV), formerly Henoch-Schönlein Purpura (HSP), is the current preferred term per the 2012 CHCC Nomenclature. It is a systemic small-vessel vasculitis characterized by the tetrad of:
- Palpable purpura
- Arthralgia/Arthritis
- Abdominal pain/GI symptoms
- Renal involvement (glomerulonephritis) — Harrison's Principles 22E (2025)
2. Epidemiology
- Most common vasculitis in children (≥75% of cases; peak age 2–8 years)
- Also occurs in adults (average age ~50 years)
- Male predominance (M:F = 1.5:1)
- Annual incidence: ~45 per million (biopsy-proven LCV overall)
- Seasonal variation: spring peak
- — Dermatology 5e; Harrison's 22E
3. Etiology & Pathogenesis
Precipitating factors:
- Upper respiratory tract infection (most common trigger — especially Group A Streptococcal pharyngitis)
- Viral infections
- Helicobacter pylori (adult and some childhood cases)
- Drugs, foods, insect bites, immunizations
Pathogenesis — Immune Complex Mediated:
- Abnormal IgA1 glycosylation → formation of IgG-IgA1 immune complexes in antigen excess
- Circulating immune complexes deposit in postcapillary venule walls
- Complement activation → neutrophil chemotaxis → endothelial injury → leukocytoclastic vasculitis
- IgA + C3 + fibrin deposition in vessel walls of both involved and uninvolved skin (pathognomonic on DIF)
- In ANCA-associated vasculitis (for contrast), damage is pauci-immune and directly neutrophil-mediated — Andrews' Diseases of the Skin; Dermatology 5e
4. Clinical Features
A. Cutaneous (100% of patients)
- Palpable purpura — hallmark; non-blanching, crops of lesions
- Distribution: extensor surfaces of lower extremities, buttocks; purpura above the waist is a marker of renal involvement
- Initially mottled; becomes hemorrhagic within 24 hours; fades in ~5 days; new crops appear over weeks
- May also show: urticarial lesions, vesicles, necrotic purpura, hemangioma-like lesions
- Preceded by mild fever, headache, joint pain, abdominal pain in ~40% of cases
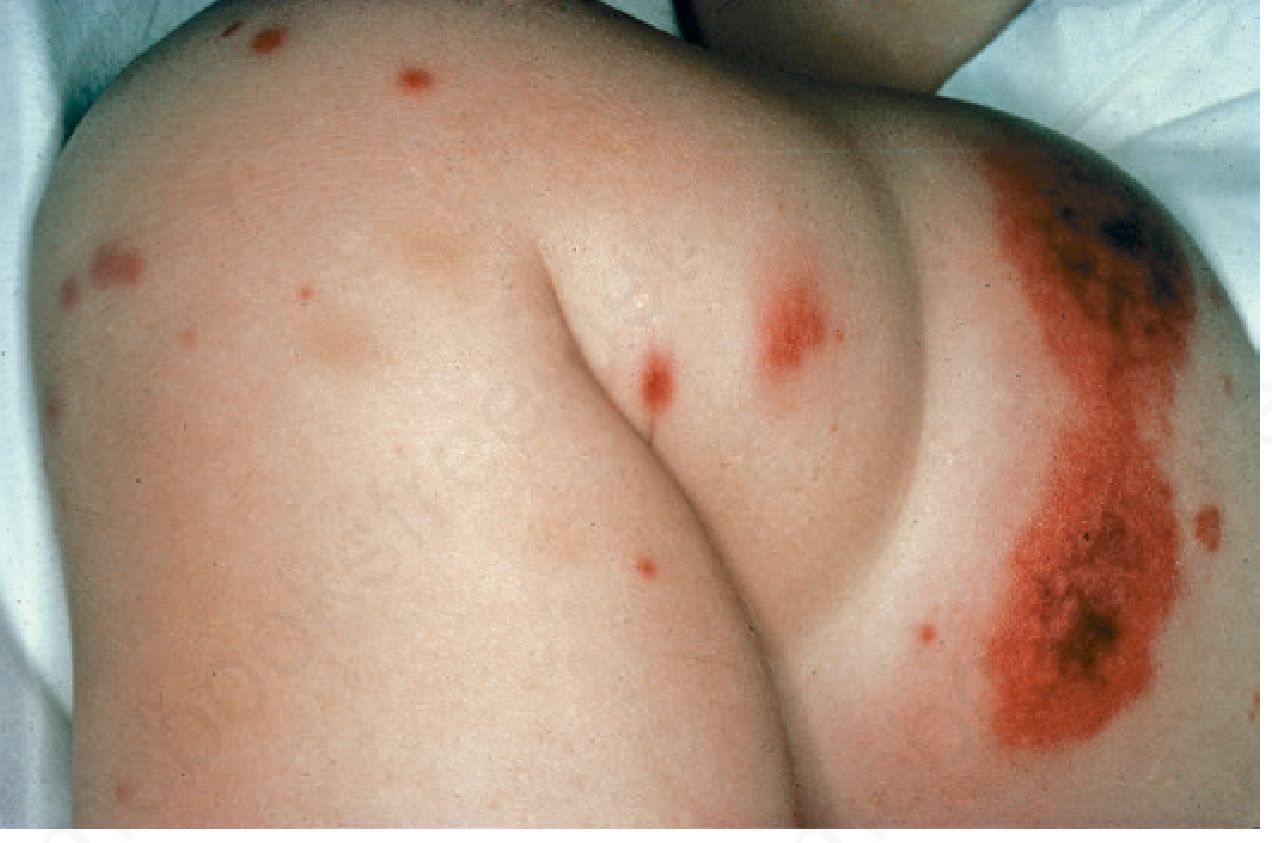
B. Joint Involvement (~63% of patients)
- Periarticular swelling of knees and ankles (most common)
- Arthralgias → frank arthritis; non-deforming, self-limiting
C. Gastrointestinal (~65–70% of patients)
- Colicky abdominal pain (most common GI symptom)
- Nausea, vomiting, diarrhea, constipation
- GI bleeding (blood and mucus per rectum)
- Bowel intussusception (most common abdominal complication in children)
- Severe pain may mimic surgical abdomen; paralytic ileus, rebound tenderness possible
- Radiograph: "spiking" or cobblestone appearance of bowel wall
D. Renal (~10–50% in children; up to 70% in adults)
- Microscopic or gross hematuria + proteinuria with RBC casts
- Usually mild glomerulonephritis — resolves spontaneously
- Adults: more severe, insidious renal course; closer follow-up required
- 1–5% of children progress to end-stage renal disease; higher rate in adults
- Baseline renal impairment + proteinuria >1 g/day + adverse biopsy findings = high risk of chronic renal disease
E. Other
- Pulmonary hemorrhage (rare but potentially fatal)
- Myocardial involvement (rare, mainly adults) — Harrison's 22E; Andrews' Diseases of the Skin; Dermatology 5e
5. Laboratory Findings
| Investigation | Finding |
|---|---|
| CBC | Mild leukocytosis; normal platelet count |
| ESR/CRP | Elevated |
| Serum IgA | Elevated in ~50% of patients |
| Serum complement (C3, C4) | Normal (distinguishes from SLE vasculitis) |
| Urinalysis | Hematuria, proteinuria, RBC casts |
| ANCA | Negative |
| Throat swab / ASO titer | To exclude streptococcal trigger |
6. Histopathology & Direct Immunofluorescence (DIF)
Light Microscopy:
- Leukocytoclastic vasculitis — neutrophilic infiltrate around post-capillary venules
- Fibrinoid necrosis of vessel walls
- Karyorrhexis ("nuclear dust"), endothelial swelling
- Extravasation of red blood cells
Direct Immunofluorescence (DIF) — PATHOGNOMONIC:
- Dominant IgA deposits in vessel walls of dermal post-capillary venules (IgA > IgG, IgM)
- Also C3 and fibrin
- Distinguishes IgAV from other LCV (which show IgG/IgM); IgA deposits persist even in older lesions (unlike other immune complexes which degrade)
- DIF of uninvolved skin can also be positive — useful when skin lesions are not present (histamine trap test) — Andrews' Diseases of the Skin; Dermatology 5e
7. Diagnosis & Diagnostic Criteria
Diagnosis is primarily clinical, supported by skin biopsy (DIF).
ACR/EULAR Classification Criteria (2022) require ≥2 of:
- Palpable purpura
- Age ≤20 at onset
- Bowel angina (colicky pain/bloody diarrhea)
- Hematuria or RBC casts on urinalysis
- IgA on biopsy (if biopsy performed)
"Histamine Trap Test": In patients with abdominal symptoms but no skin lesions — histamine injected intradermally → skin biopsied at 4 hours → identifies IgA in vessel walls → confirms diagnosis.
Renal biopsy: Rarely needed for diagnosis; may give prognostic information — shows mesangial IgA deposits (identical pattern to IgA nephropathy/Berger's disease).
8. Differential Diagnosis
| Condition | Differentiating Feature |
|---|---|
| Other LCV (idiopathic/drug) | IgG/IgM dominant on DIF; no IgA |
| Meningococcemia | Stellate/retiform purpura, fever, meningism, DIC |
| ITP | Thrombocytopenic, non-palpable purpura |
| Cryoglobulinemic vasculitis | Low C4, mixed cryoglobulins, often HCV-associated |
| Acute hemorrhagic edema of infancy | Age <2 years, target-like lesions, DIF may show IgM |
| Child abuse (Battered child) | Distribution pattern, exclusion of vasculitis on biopsy |
9. Treatment
Self-limiting in most children — supportive care suffices.
| Clinical Scenario | Treatment |
|---|---|
| Skin purpura alone | Supportive; bed rest, elevation of legs; NSAIDs for arthralgia |
| Arthralgia / tissue edema | Prednisone 1 mg/kg/day — reduces edema, arthralgia, abdominal pain; taper per response |
| Severe abdominal pain / GI bleeding | Short course IV corticosteroids |
| Severe / progressive glomerulonephritis | Corticosteroids + immunosuppressive agent (azathioprine, cyclophosphamide, or mycophenolate mofetil) |
| H. pylori-positive adults with GI symptoms | Eradication therapy |
| Refractory / crescentic GN | ACE inhibitors/ARBs; plasma exchange; rituximab (emerging evidence — PMID 39249581) |
Note: Steroids do not prevent or shorten renal disease, do not prevent recurrences, and do not shorten overall disease duration. — Harrison's 22E; Andrews' Diseases of the Skin
10. Prognosis & Complications
- Excellent overall prognosis in children
- Mortality: extremely rare
- 1–5% of children → ESRD; higher rate in adults
- Relapse rate: 10–40% of patients (usually within weeks to months of initial episode)
- Poor prognostic indicators:
- Purpura above the waist
- Persistent proteinuria (>1 g/day)
- Crescentic glomerulonephritis on renal biopsy
- Adult onset
- Presence of IgM in lesional skin (indicator of renal involvement)
Summary Box (Quick Revision)
| Feature | Key Point |
|---|---|
| Type | IgA-mediated immune complex small vessel vasculitis |
| Age | Children 2–8 years (most common); adults also |
| Tetrad | Purpura + Arthritis + Abdomen + Renal |
| Trigger | URTI / Streptococcal pharyngitis |
| Pathognomonic | IgA deposits on DIF (skin + kidney) |
| Complement | Normal (differentiates from SLE) |
| IgA serum | Elevated in 50% |
| GI complication | Intussusception |
| Treatment | Supportive; steroids for systemic symptoms; IS agents for GN |
| Prognosis | Excellent in children; guarded in adults with renal involvement |
Sources: Dermatology 2-Volume Set 5e (Bolt/Griffiths), Andrews' Diseases of the Skin — Clinical Dermatology, Harrison's Principles of Internal Medicine 22E (2025), Fitzpatrick's Dermatology; recent systematic review on rituximab in IgA vasculitis nephritis (PMID 39249581, Clin Exp Med 2024).
Classify porphyrias.Describe heme biosynthesis and comment on PCT in 10marks format for 3rd yr dermatology post graduate theory examination
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
porphyria cutanea tarda treatment
Loading Image
Reading File
I now have all the comprehensive material needed. Let me compile the complete, structured 10-mark answer:
Classification of Porphyrias, Heme Biosynthesis & Porphyria Cutanea Tarda
PART I: HEME BIOSYNTHESIS
Heme is an iron-containing tetrapyrrole essential for hemoglobin, myoglobin, respiratory cytochromes, and cytochrome P450 enzymes. ~85% of daily heme synthesis occurs in erythroid precursor cells (for hemoglobin); hepatocytes account for most of the remainder (primarily for CYP enzymes).
The pathway involves 8 enzymatic steps converting glycine + succinyl-CoA → heme, encoded by 9 genes (ALAS has two distinct genes: ALAS1 for housekeeping; ALAS2 for erythroid-specific).
Compartments
- Mitochondria: Steps 1, 6, 7, 8 (first and last three enzymes)
- Cytosol: Steps 2–5 (middle four enzymes)
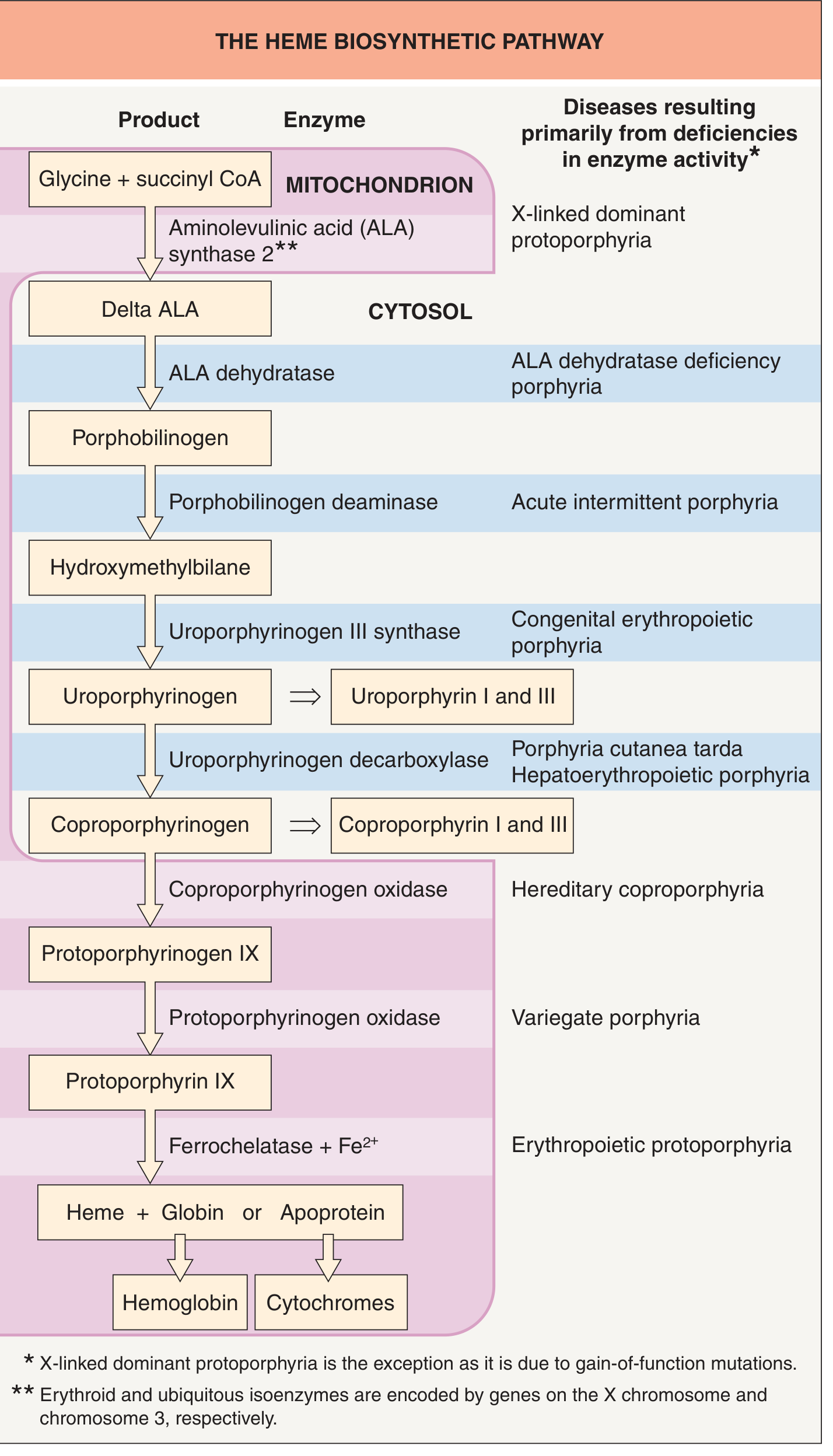
The Eight Enzymatic Steps
| Step | Location | Substrate | Enzyme | Product | Porphyria if Deficient |
|---|---|---|---|---|---|
| 1 | Mitochondria | Glycine + Succinyl-CoA | ALA Synthase (ALAS1/ALAS2) | δ-Aminolevulinic acid (ALA) | X-linked dominant protoporphyria (gain-of-function ALAS2) |
| 2 | Cytosol | 2× ALA | ALA Dehydratase (zinc-dependent) | Porphobilinogen (PBG) | ALA-dehydratase deficiency porphyria (ALADP) |
| 3 | Cytosol | 4× PBG | HMB Synthase (PBG deaminase) | Hydroxymethylbilane (HMB) | Acute Intermittent Porphyria (AIP) |
| 4 | Cytosol | HMB | Uroporphyrinogen III Synthase | Uroporphyrinogen III | Congenital Erythropoietic Porphyria (CEP) |
| 5 | Cytosol | Uroporphyrinogen III | Uroporphyrinogen Decarboxylase (UROD) | Coproporphyrinogen III | PCT & Hepatoerythropoietic porphyria (HEP) |
| 6 | Mitochondria | Coproporphyrinogen III | Coproporphyrinogen Oxidase | Protoporphyrinogen IX | Hereditary Coproporphyria (HCP) |
| 7 | Mitochondria | Protoporphyrinogen IX | Protoporphyrinogen Oxidase (PPOX) | Protoporphyrin IX | Variegate Porphyria (VP) |
| 8 | Mitochondria | Protoporphyrin IX + Fe²⁺ | Ferrochelatase | HEME | Erythropoietic Protoporphyria (EPP) |
Key regulatory point: ALAS1 (hepatic) is the rate-limiting step, induced by drugs, steroids, fasting, and PGC-1α. Heme exerts negative feedback on ALAS1 — when downstream enzyme is deficient, heme falls → ALAS1 upregulated → precursors accumulate. — Harrison's Principles 22E; Dermatology 5e
PART II: CLASSIFICATION OF PORPHYRIAS
A. By Site of Principal Enzyme Expression (Traditional)
| Category | Disorders |
|---|---|
| Hepatic | AIP, ALADP, VP, HCP, PCT (types I, II, III) |
| Erythropoietic | CEP (Günther disease), EPP, X-linked dominant protoporphyria (XLP) |
| Mixed/Hepatoerythropoietic | HEP (homozygous PCT) |
B. By Clinical Presentation — ACUTE vs NON-ACUTE (Clinically most useful)
Acute (Neurovisceral) Porphyrias
| Disorder | Gene | Inheritance | Enzyme Deficient | Skin? | Key Features |
|---|---|---|---|---|---|
| Acute Intermittent Porphyria (AIP) | HMBS | AD | HMB synthase (PBG deaminase) | No | Most common acute porphyria; neurovisceral attacks only |
| Variegate Porphyria (VP) | PPOX | AD | Protoporphyrinogen oxidase | Yes | Neurocutaneous; skin like PCT + acute attacks; common in South Africa |
| Hereditary Coproporphyria (HCP) | CPOX | AD | Coproporphyrinogen oxidase | Sometimes | Acute attacks + occasional blistering |
| ALA-Dehydratase Deficiency Porphyria (ALADP) | ALAD | AR | ALA dehydratase | No | Extremely rare (<10 cases); neurologic only |
Non-Acute Porphyrias (All Cutaneous)
| Disorder | Gene | Inheritance | Enzyme Deficient | Onset | Skin Features |
|---|---|---|---|---|---|
| PCT (Types I, II, III) | UROD | Type I: Acquired; Type II: AD; Type III: AD | Uroporphyrinogen decarboxylase | 3rd–4th decade | Photosensitivity, skin fragility, bullae, milia, hypertrichosis, hyperpigmentation, sclerodermoid changes |
| Erythropoietic Protoporphyria (EPP) | FECH | AD/AR | Ferrochelatase ↓ | Infancy/childhood | Burning, erythema, edema, waxy scars — no blistering; photosensitivity immediate |
| X-linked dominant Protoporphyria (XLP) | ALAS2 | X-linked dominant | ALAS2 gain-of-function | Childhood | Clinically similar to EPP |
| Congenital Erythropoietic Porphyria (CEP) | UROS | AR | Uroporphyrinogen III synthase | Infancy | Severe; mutilating scarring, erythrodontia (pink teeth), hemolytic anemia, porphyrinuria (pink diapers) |
| Hepatoerythropoietic Porphyria (HEP) | UROD | AR | Uroporphyrinogen decarboxylase (homozygous) | Infancy | Homozygous PCT; CEP-like features |
C. By Skin Manifestations — Dermatologist's Classification (Dermatology 5e)
| Cutaneous Porphyrias | Non-Cutaneous Porphyrias |
|---|---|
| PCT | AIP |
| EPP | ALADP |
| VP | |
| HCP | |
| CEP | |
| HEP | |
| XLP |
Mnemonic for cutaneous porphyrias: PCT, EPP, VP, HCP, CEP, HEP, XLP → "PEVHCHX"
D. By Biochemical Pattern (Fluorescence Emission)
| Porphyria | Plasma fluorescence peak | Key urine/stool finding |
|---|---|---|
| PCT | — | ↑↑ Uroporphyrins + heptacarboxylate porphyrins in urine |
| VP | 624–626 nm | ↑↑ Fecal proto + coproporphyrins |
| HCP | 619 nm | ↑↑ Fecal coproporphyrins |
| AIP | — | ↑↑ Urine ALA + PBG (normal in remission) |
PART III: PORPHYRIA CUTANEA TARDA (PCT) — DETAILED DISCUSSION
1. Definition & Overview
PCT is the most common porphyria worldwide. It results from a deficiency of uroporphyrinogen decarboxylase (UROD) — the 5th enzyme in heme biosynthesis — leading to accumulation of uroporphyrins and heptacarboxylate porphyrins in the liver, plasma, skin, and urine.
— Dermatology 5e; Harrison's 22E (2025)
2. Types of PCT
| Type | UROD defect location | Genetics | Frequency |
|---|---|---|---|
| Type I (Sporadic/Acquired) | Liver only (acquired inhibition) | No UROD mutation | ~80% of cases |
| Type II (Familial/Hereditary) | All tissues (50% of normal) | AD — UROD gene mutation (heterozygous) | ~20% of cases |
| Type III (Familial, normal RBC UROD) | Unclear | AD — no detectable UROD mutation in RBC | Rare |
| HEP | All tissues (<10% of normal) | AR — homozygous UROD mutations | Extremely rare |
For clinical symptoms to manifest, UROD activity must fall to ≤20% of normal — hence even type II carriers require additional susceptibility factors.
3. Pathogenesis
- UROD inhibition → uroporphyrinogen III and hepta-carboxylate porphyrinogens accumulate → auto-oxidize to photoactive uroporphyrins
- Requires iron + oxidative stress in the liver → generation of a uroporphomethene (UROD inhibitor)
- Uroporphyrins are water-soluble → deposit in skin dermis and papillary vessels
- Soret band absorption (400–410 nm, UVA/visible light) → singlet oxygen + free radicals → lipid peroxidation + protein cross-linking → subepidermal blister formation
- Festooning of dermal papillae due to PAS-positive material deposition
4. Precipitating / Susceptibility Factors
| Factor | Mechanism |
|---|---|
| Hepatitis C virus (HCV) | Most common trigger; promotes hepatic iron overload + oxidative damage |
| Alcohol | Long-recognized; hepatotoxic + increases iron |
| Hemochromatosis (HFE mutations) | C282Y and H63D mutations increase hepatic iron — synergistic |
| Estrogens (OCP, HRT) | Promote iron accumulation in liver |
| HIV | Independent risk factor |
| Smoking (>10 cigs/day) | Earlier onset by ~1 decade |
| Hexachlorobenzene / dioxins | Chemical induction (Turkish epidemic, 1950s) |
| Type 2 Diabetes / Metabolic syndrome | NASH → hepatic iron dysregulation |
| Lupus erythematosus | Frequently co-reported with PCT |
5. Clinical Features
A. Cutaneous (Pathognomonic)
- Skin fragility and blistering on sun-exposed areas: dorsal hands and forearms are most commonly affected; also face, ears, dorsal feet (especially shins in women), legs
- Bullae are non-inflammatory, thin-roofed, rupture easily → erosions → shallow ulcers → heal with atrophic scars + milia
- Hypertrichosis: temporal cheeks, temples (especially in women) — characteristic feature
- Hyperpigmentation: face, neck, hands — brownish discoloration
- Violaceous/pink tint of periorbital area and neck (heliotrope-like)
- Sclerodermoid/morpheiform changes: back of neck, preauricular areas, thorax, fingers, scalp → may cause scarring alopecia — directly correlates with urine uroporphyrin levels
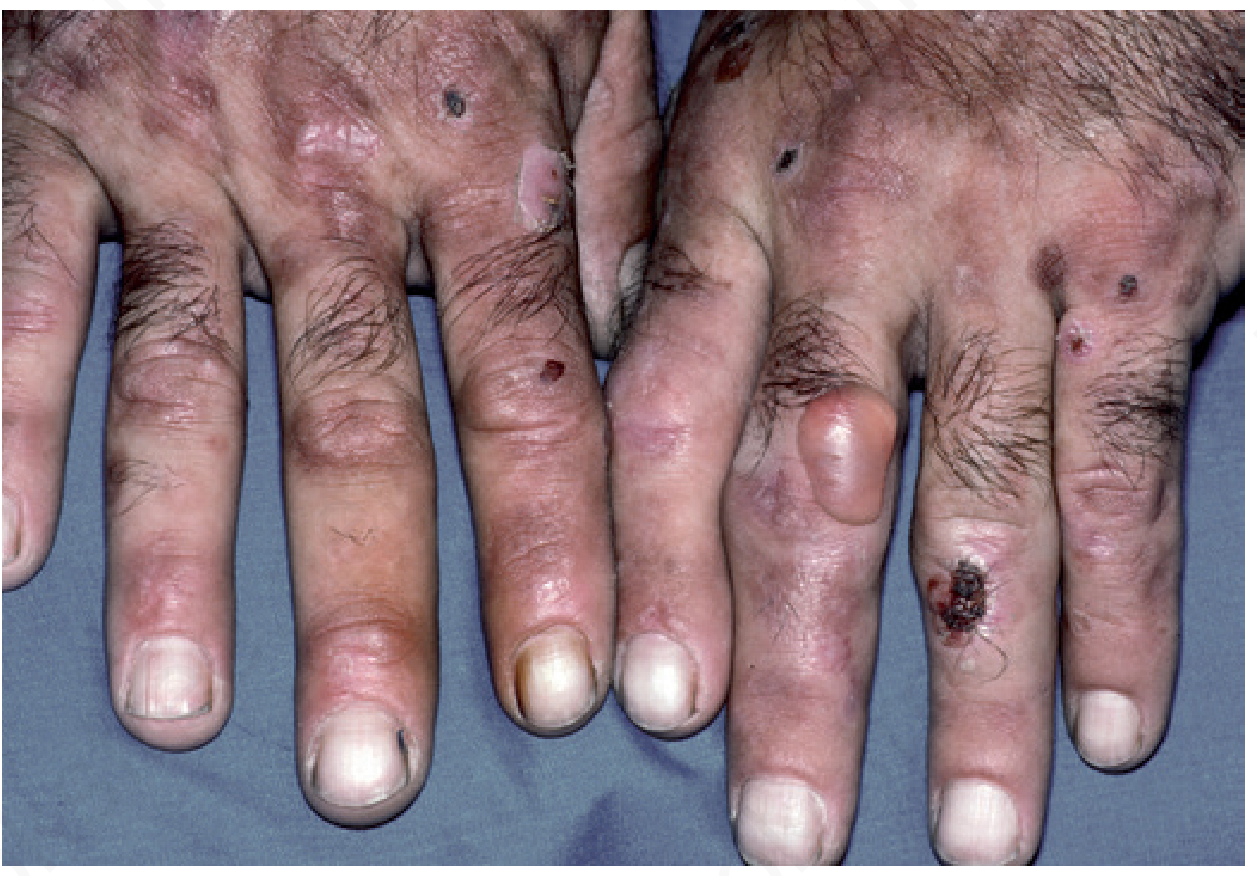
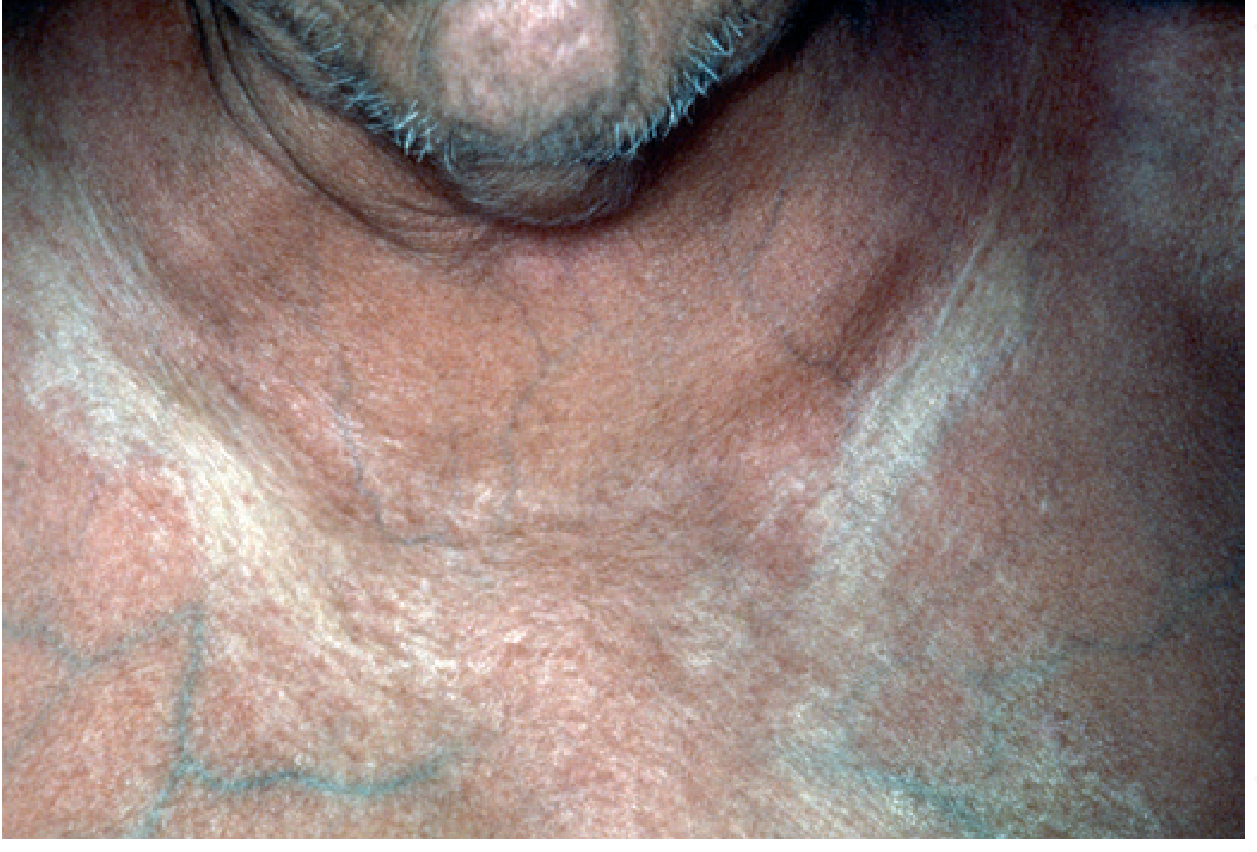
B. Systemic
- Liver disease frequently present: hepatitis C, alcoholic liver disease, steatohepatitis
- Risk of hepatocellular carcinoma (3.5× increased risk)
- Neurological features are ABSENT (unlike AIP/VP) — key distinguishing point
6. Investigations / Diagnosis
| Test | Finding in PCT |
|---|---|
| Urine porphyrins | ↑↑↑ Uroporphyrins + heptacarboxylate porphyrins (diagnostic) |
| Urine ALA | Mildly elevated |
| Urine PBG | Normal (differentiates from AIP) |
| Fecal porphyrins | ↑ Isocoproporphyrins (diagnostic for UROD deficiency) |
| Plasma porphyrins | Elevated; fluorescence at neutral pH |
| RBC UROD activity | ↓ in Type II (50% of normal); normal in Type I |
| Serum ferritin | Elevated (iron overload) |
| Wood's lamp (urine) | Coral-pink fluorescence (uroporphyrins) |
| LFTs + HCV serology | Often abnormal |
| HFE mutations (C282Y, H63D) | Increased frequency in PCT |
Plasma fluorescence scan (diluted plasma at neutral pH): differentiates PCT (no specific peak) from VP (624–626 nm) and HCP (619 nm).
7. Histopathology
- Subepidermal cell-poor blister (minimal inflammatory infiltrate)
- Festooning of dermal papillae — papillae project upward into blister floor (characteristic)
- PAS-positive material deposition around dermal vessels
- Thickened dermal vessel walls
- Direct Immunofluorescence (DIF): Immunoglobulins (IgG) + complement (C3) around blood vessels in upper dermis — "doughnut pattern"
8. Differential Diagnosis
| Condition | Differentiation |
|---|---|
| Pseudoporphyria (NSAIDs — naproxen most common; voriconazole; tetracyclines; dialysis) | Identical clinically but normal porphyrins; no hypertrichosis/dyspigmentation/sclerosis |
| Variegate Porphyria (VP) | Skin identical to PCT but also has acute attacks; ↑↑ fecal protoporphyrins; plasma fluorescence 624 nm |
| Epidermolysis bullosa acquisita | DIF shows IgG at DEJ (linear); no porphyrins |
| CEP | Onset in infancy; erythrodontia; severe scarring |
| Drug-induced phototoxicity | History of drug, photodistributed, no porphyrins |
9. Treatment
Goal: Reduce porphyrin load by depleting hepatic iron and promoting porphyrin excretion.
First-line: Phlebotomy (Venesection)
- Remove 450 mL blood every 1–2 weeks
- Target: serum ferritin → lower limit of normal
- Usually requires 5–6 sessions (more in hemochromatosis-associated PCT)
- Monitor Hb and ferritin to avoid iatrogenic iron deficiency anemia
- Plasma porphyrins normalize after target ferritin reached
- Most effective, cheapest, safest treatment
Second-line: Low-dose Antimalarials
- Chloroquine phosphate 125 mg twice weekly OR Hydroxychloroquine 200 mg twice weekly
- Mechanism: complex with porphyrins → mobilize from hepatocytes → promote urinary excretion
- Do NOT use standard doses — risk of transient severe photosensitivity flare + hepatocellular damage
- Hydroxychloroquine equally effective and safe as phlebotomy (studies confirm)
- Preferred when phlebotomy is contraindicated (anemia, cardiovascular disease)
Specific Situations
| Situation | Treatment |
|---|---|
| PCT + HCV infection | Direct-acting antivirals (DAAs) for HCV — clearance of HCV can lead to complete PCT remission (first-line in HCV-positive PCT) |
| PCT + End-stage renal disease (anuric patients) | Erythropoietin → stimulates erythropoiesis → reduces iron load; phlebotomy difficult |
| PCT + Hemochromatosis | More phlebotomies needed; may also require chelation |
| All patients | Avoid/eliminate precipitating factors: alcohol, estrogens, iron supplements, offending drugs; sun protection (UVA-blocking sunscreens, protective clothing) |
10. Prognosis
- Complete remission achievable in nearly all patients with appropriate therapy
- Relapse possible — monitor plasma porphyrins every 6–12 months
- Risk of HCC requires periodic hepatic surveillance
- Familial (Type II) PCT patients: genetic counseling for family members
- HEP (homozygous) PCT: severe childhood-onset disease; may require bone marrow transplantation (as for CEP)
Summary Box — PCT at a Glance
| Feature | Key Point |
|---|---|
| Enzyme deficient | Uroporphyrinogen decarboxylase (UROD) — enzyme 5 |
| Most common porphyria | Worldwide |
| Key substrate accumulating | Uroporphyrins + heptacarboxylate porphyrins |
| Hallmark skin lesion | Skin fragility + non-inflammatory subepidermal bullae on sun-exposed areas |
| Diagnostic triad | Blistering + hypertrichosis + hyperpigmentation |
| Neurological features | Absent |
| Wood's lamp urine | Coral-pink fluorescence |
| Precipitants | HCV, alcohol, estrogens, hemochromatosis (HFE mutations), HIV |
| First-line treatment | Phlebotomy |
| Second-line | Low-dose chloroquine/hydroxychloroquine |
| HCV + PCT | Direct-acting antivirals |
Sources: Dermatology 2-Volume Set 5e (Bolt/Griffiths), Andrews' Diseases of the Skin — Clinical Dermatology, Harrison's Principles of Internal Medicine 22E (2025), Fitzpatrick's Dermatology. For recent evidence on PCT management, see Sarkany & Phillips (Liver Int 2024, PMID 38813949) and Dickey et al. — Update on the Porphyrias (Annu Rev Med 2024, PMID 37540847).
classify photodermatoses.Discuss PMLE in 10markd for 3rd yr dermatology post graduate theory examination
Finding Sources
Finding Sources
Searching PubMed
polymorphic light eruption pathogenesis treatment
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.