Treatment of nephritic syndrome
Reading File
Finding Sources
Searching PubMed
"nephritic syndrome" AND treatment
Searching the Web
nephritic syndrome treatment management 2025 guidelines
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Finding Sources
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Treatment of Nephritic Syndrome
Nephritic syndrome is characterized by glomerular hematuria (dysmorphic RBCs or RBC casts), hypertension, edema, oliguria/reduced GFR, and non-nephrotic proteinuria. Management is divided into two components: (1) general/supportive care applicable to all causes, and (2) disease-specific immunosuppressive therapy targeted to the underlying etiology.
I. General Supportive Management
These measures apply regardless of the underlying cause:
1. Fluid and Sodium Management
- Dietary sodium restriction (60–80 mmol/24 h) combined with loop diuretics (e.g., furosemide) is the mainstay for edema control.
- Aim for fluid removal of no more than 1–2 kg/day in adults to avoid hypovolemia.
- In diuretic resistance: combine a loop diuretic with a thiazide or metolazone, or add amiloride (nephritic syndrome activates ENaC).
- In severe hypoalbuminemia, GI absorption of oral diuretics may be impaired — IV bolus or infusion may be needed.
2. Blood Pressure Control
- ACE inhibitors or ARBs are first-line — they reduce proteinuria and slow GFR decline.
- Target BP <130/80 mmHg (or lower if proteinuria is significant).
- NSAIDs should be avoided — they risk precipitating AKI, especially in the setting of pre-existing kidney impairment.
3. Dietary Protein
- Ensure adequate protein intake of 0.8–1 g/kg/day with high carbohydrate to minimize catabolism.
4. Lipid Management
- Statins or statin/ezetimibe combinations in adults >50 years with CKD stage 3–5, or younger adults with significant comorbidity.
5. Dialysis
- Indicated if severe, rapidly progressive renal failure (RPGN pattern) — serves as bridge while awaiting immunosuppressive response.
6. Avoidance of Nephrotoxins
- Caution with radiocontrast agents, aminoglycosides, and PPIs.
II. Disease-Specific Treatment
Nephritic syndrome has several distinct causes — each requires targeted therapy.
A. Post-Infectious (Post-Streptococcal) GN
The most common classic cause, particularly in children.
- Treatment is supportive — control hypertension, manage edema, dialysis if needed.
- Antibiotics (e.g., penicillin) for active streptococcal infection in the patient and close contacts — but antibiotics do not reduce the occurrence of nephritis once established.
- No role for immunosuppressive therapy, even in the setting of crescents on biopsy.
- Prognosis in children is excellent; most cases resolve within weeks.
- Renal biopsy is not required unless atypical features are present.
Harrison's Principles of Internal Medicine 22E, 2025
B. IgA Nephropathy (IgAN)
The most common primary GN worldwide. Treatment is indicated for proteinuria >0.5 g/day.
Step 1: RAAS Blockade (All patients with proteinuria >500 mg/day)
- ARBs at escalating doses (e.g., losartan up to 200 mg/day) — shown superior to standard dosing for reducing proteinuria and preserving GFR.
- Target urinary protein <1 g/day.
- Dietary sodium restriction enhances effect.
Step 2: Glucocorticoids (if proteinuria persists >1–3.5 g/day despite RAAS blockade, with preserved GFR)
- 6-month course of IV + oral glucocorticoids — associated with reduced risk of doubling creatinine and improved renal survival (97% vs. 53% placebo at 10 years in one landmark RCT).
- Not beneficial if significant CKD/interstitial fibrosis is already present (STOP-IgAN, TESTING trials raised concerns about toxicity vs. benefit in CKD patients).
Step 3: Combined Immunosuppression (Progressive disease)
- Cytotoxic therapy (e.g., cyclophosphamide) reserved for documented progression despite above.
- Novel therapy: Targeted-release budesonide (Nefecon) — a KDIGO 2025-recognized option delivering corticosteroid to gut-associated lymphoid tissue, reducing galactose-deficient IgA1 production with less systemic toxicity.
- SGLT2 inhibitors (e.g., dapagliflozin) reduce proteinuria and slow progression; now incorporated in recent KDIGO guidance.
- Sparsentan (dual endothelin/angiotensin receptor blocker) approved for IgAN.
Brenner & Rector's The Kidney; Comprehensive Clinical Nephrology 7e; KDIGO 2025 IgAN Guideline
C. Lupus Nephritis (Class III/IV — Proliferative)
Class III (focal) and IV (diffuse) present with nephritic syndrome and are treated aggressively.
Induction Therapy
-
High-dose corticosteroids (IV methylprednisolone pulses 500–1000 mg × 3 days, then oral prednisone 0.5–1 mg/kg/day tapered) PLUS one of:
- Mycophenolate mofetil (MMF) 2–3 g/day — preferred in many centers (equivalent efficacy to cyclophosphamide, lower gonadotoxicity)
- IV Cyclophosphamide (low-dose NIH or Euro-Lupus regimen)
-
Triple therapy options now recommended by KDIGO/EULAR 2024:
- Voclosporin (CNI) + MMF + glucocorticoids — AURORA-1 trial demonstrated superior complete renal response
- Belimumab (anti-BLyS) + MMF/cyclophosphamide + glucocorticoids — BLISS-LN trial showed benefit
- Hydroxychloroquine should be continued in all patients with lupus unless contraindicated.
Maintenance Therapy
- MMF 1–2 g/day (preferred) or azathioprine 1.5–2 mg/kg/day for at least 3–5 years.
- Continue hydroxychloroquine.
- RAS blockade for proteinuria reduction.
Harrison's Principles of Internal Medicine 22E; Comprehensive Clinical Nephrology 7e; NKF Primer 8e
D. ANCA-Associated Vasculitis (AAV) — Pauci-Immune GN
Includes granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
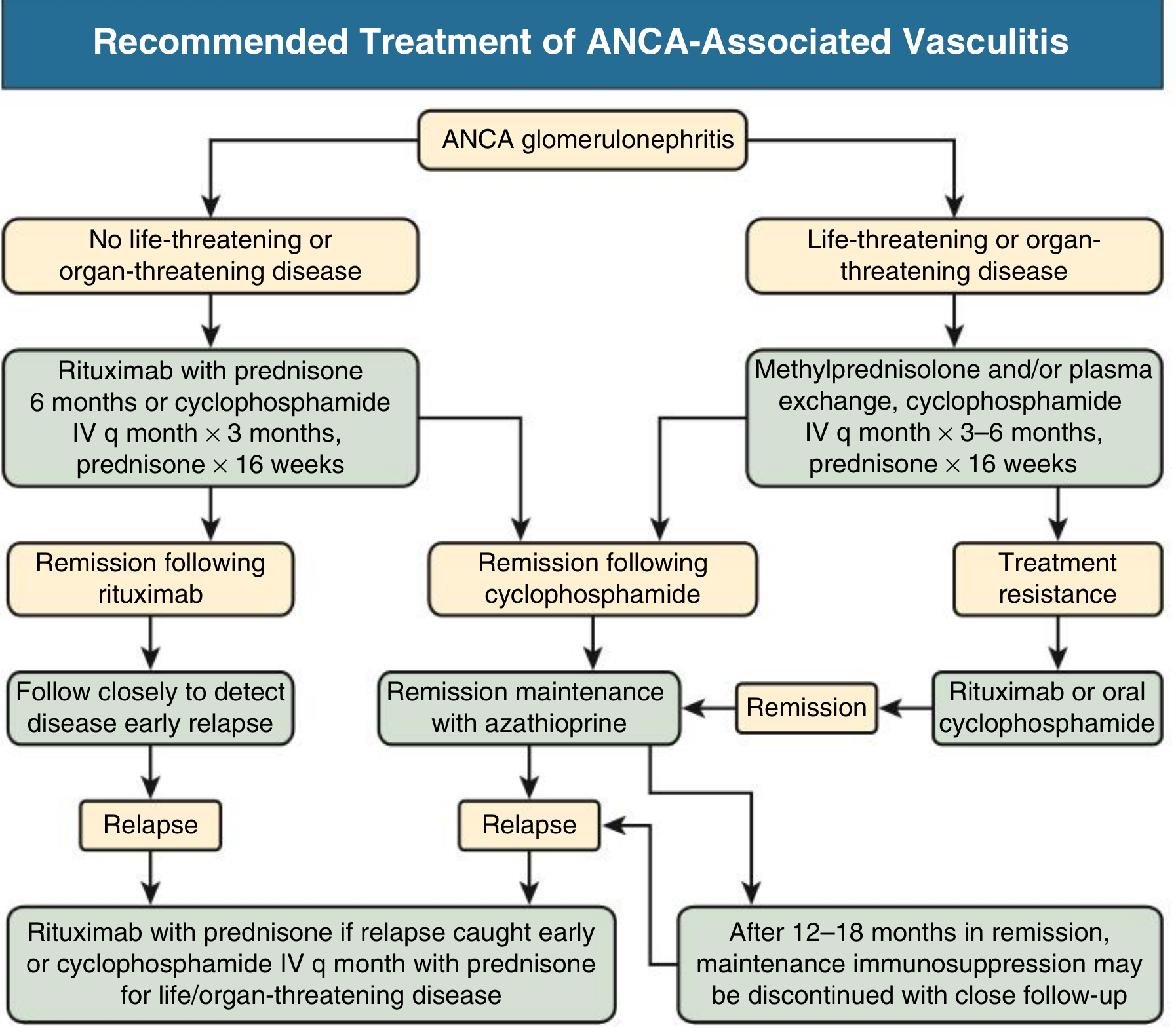
Induction Therapy
Non-life/organ-threatening disease:
- Rituximab (375 mg/m² × 4 doses or 1g × 2 doses 14 days apart) + prednisone × 16 weeks
- Alternative: IV cyclophosphamide monthly × 3 months + prednisone × 16 weeks
Life/organ-threatening disease (severe kidney involvement, pulmonary hemorrhage):
- IV methylprednisolone pulses + IV cyclophosphamide monthly × 3–6 months + prednisone × 16 weeks
- Combination IV cyclophosphamide + rituximab reduces mortality, relapse, and ESKD vs. either agent alone
Plasma exchange (PLEX):
- PEXIVAS trial (2020) showed no overall reduction in mortality or ESKD with PLEX
- Still considered in: eGFR <30 mL/min without significant chronic fibrosis, or severe pulmonary hemorrhage requiring mechanical ventilation/O₂ supplementation
- NOT recommended as routine for all severe presentations
Rituximab vs. cyclophosphamide: RAVE and RITUXVAS trials showed equivalent remission rates; rituximab is more effective in PR3-ANCA (vs. MPO-ANCA) disease.
Maintenance Therapy
- After cyclophosphamide induction: switch to azathioprine at remission (12–18 months)
- After rituximab induction: close follow-up for early relapse; re-treat with rituximab at relapse
- Maintenance immunosuppression may be discontinued after 12–18 months of sustained remission
Comprehensive Clinical Nephrology 7e
E. Anti-GBM Antibody Disease (Goodpasture Syndrome)
A rapidly progressive, potentially fatal GN.
Treatment
- IV methylprednisolone pulses (1 g/day × 3 days) → oral prednisolone 1 mg/kg/day
- Cyclophosphamide (age- and GFR-adjusted oral or IV)
- Plasma exchange — mandatory and urgent; 4L exchanges daily or every other day × 14 days or until anti-GBM antibodies are undetectable. Particularly vital with pulmonary hemorrhage.
Prognostic thresholds:
- Serum creatinine <5.0 mg/dL at presentation → 90% kidney survival at 5 years
- Dialysis-dependent at presentation or 100% circumferential crescents → unlikely renal recovery; immunosuppression only indicated if concomitant pulmonary hemorrhage
- Transplantation after antibody disappearance — recurrence is rare
NKF Primer 8e; Comprehensive Clinical Nephrology 7e
F. Rapidly Progressive GN (RPGN) — General Approach
When crescentic GN causes rapid deterioration over days–weeks regardless of underlying cause:
| Step | Treatment |
|---|---|
| 1 | IV methylprednisolone 500–1000 mg/day × 3 days (pulse) |
| 2 | Oral prednisolone 1 mg/kg/day with taper |
| 3 | Add cyclophosphamide or rituximab based on etiology |
| 4 | Plasma exchange if anti-GBM or severe AAV with pulmonary hemorrhage |
| 5 | Renal replacement therapy (dialysis) as bridge if needed |
III. Summary Table
| Etiology | First-Line Immunosuppression | Key Points |
|---|---|---|
| Post-infectious GN | None (supportive only) | Antibiotics for active infection; self-limiting |
| IgA nephropathy | RAAS blockade → glucocorticoids | Novel: sparsentan, budesonide, SGLT2i |
| Lupus nephritis (III/IV) | Steroids + MMF or CYC | Triple therapy: ± voclosporin or belimumab |
| ANCA-associated vasculitis | Steroids + rituximab or CYC | PLEX only for severe pulmonary hemorrhage |
| Anti-GBM disease | Steroids + CYC + PLEX | Urgency paramount; poor prognosis if dialysis-dependent |
| RPGN (crescentic) | Pulse steroids + CYC/rituximab | Etiology determines maintenance |
Sources: Comprehensive Clinical Nephrology 7e · NKF Primer on Kidney Diseases 8e · Harrison's Principles of Internal Medicine 22E · Brenner & Rector's The Kidney · KDIGO 2021 Glomerular Diseases Guideline · KDIGO 2025 IgAN Guideline
This is a shared conversation. Sign in to Orris to start your own chat.