Pathogenesis of atherosclerosis
Reading File
Finding Sources
Searching PubMed
"atherosclerosis"[MeSH Terms] AND "pathogenesis"
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I have all the essential content needed for a thorough, well-illustrated answer. Let me now compose the comprehensive response.
Pathogenesis of Atherosclerosis
The core concept is the "response to injury" hypothesis: atherosclerosis is a chronic inflammatory response of the arterial wall to endothelial injury, driven by the interaction of lipids, immune cells, and vascular smooth muscle cells (SMCs).
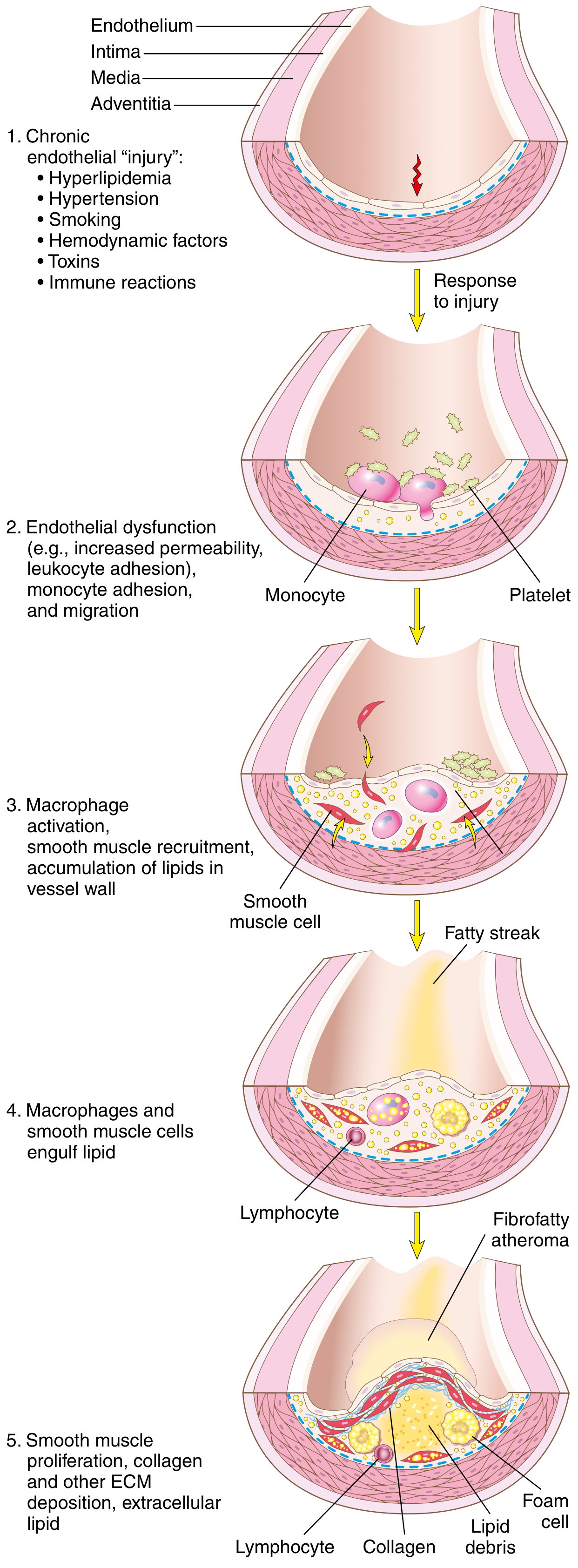
Step 1 - Endothelial Injury and Dysfunction
Endothelial cell (EC) injury is the cornerstone of atherogenesis. Importantly, early atherosclerotic lesions begin at sites of intact but dysfunctional endothelium - frank denudation is not required.
Causes of EC dysfunction:
- Hemodynamic disturbances (turbulent flow at branch points, ostia, posterior abdominal aorta wall)
- Hyperlipidemia / hypercholesterolemia (the two most important causes)
- Hypertension
- Cigarette smoke toxins (oxidative stress, NO suppression)
- Inflammatory cytokines (e.g., TNF)
- Diabetes-associated advanced glycation end-products
- Infectious agents (e.g., Chlamydia pneumoniae, CMV)
Dysfunctional ECs exhibit:
- Increased permeability - allowing lipoprotein entry into the intima
- Upregulation of adhesion molecules (VCAM-1, ICAM-1, selectins) - promoting leukocyte adhesion
- Enhanced thrombogenicity
- Reduced NO bioavailability - impairing vasodilation and enabling SMC proliferation
Hemodynamic explanation for why plaques form at predictable sites: non-turbulent laminar flow induces "atheroprotective" genes in ECs, while turbulent flow at branch points and bends suppresses them.
Step 2 - Lipoprotein Accumulation and Oxidation
Increased LDL permeates the dysfunctional endothelium and accumulates in the intima. Evidence for the central role of lipids:
- Plaques are dominated by cholesterol and cholesterol esters
- Genetic LDL receptor defects (familial hypercholesterolemia) cause accelerated atherosclerosis
- Diets high in cholesterol produce atherosclerosis in experimental animals
- Reducing LDL dramatically decreases coronary events clinically
Once in the intima, LDL undergoes oxidative modification (oxLDL) - the most atherogenic form. oxLDL:
- Stimulates ECs and macrophages to release cytokines and growth factors
- Is chemotactic for monocytes
- Is taken up avidly by macrophage scavenger receptors (not the normal LDL receptor), driving foam cell formation
- Is directly cytotoxic to ECs and SMCs
HDL is atheroprotective because it promotes reverse cholesterol transport (removing cholesterol from the arterial wall) and has antioxidant properties.
Step 3 - Monocyte Recruitment and Foam Cell Formation
- Dysfunctional ECs upregulate VCAM-1 and selectins, causing monocytes and T lymphocytes to roll, adhere, and transmigrate into the intima.
- Chemokines (e.g., MCP-1/CCL2) produced by ECs and SMCs attract monocytes into the sub-endothelial space.
- Monocytes differentiate into macrophages, activated by cytokines and oxLDL.
- Activated macrophages engulf oxLDL via scavenger receptors (SR-A, CD36) - a process that is unregulated (unlike the downregulatable LDL receptor) - leading to massive lipid accumulation.
- Lipid-engorged macrophages become foam cells, which cluster under the intima to form the earliest visible lesion: the fatty streak.
Fatty streaks appear as yellow intimal lipid deposits. They are found in the aortas of infants younger than 1 year and in virtually all children older than 10 years. Not all fatty streaks progress to plaques, but coronary fatty streaks in adolescents form at the same anatomic sites prone to plaques later in life.
Step 4 - SMC Recruitment, Proliferation, and ECM Deposition
Growth factors and cytokines released by activated macrophages, platelets, and ECs (especially PDGF, FGF, TGF-β) stimulate:
- Migration of SMCs from the tunica media (or from circulating precursors) into the intima
- Phenotypic change of SMCs from "contractile" to "synthetic" phenotype
- Proliferation of intimal SMCs
- ECM synthesis - collagen, elastic fibers, proteoglycans - forming the fibrous cap
Lipids accumulate both extracellularly and within macrophages and SMCs (creating more foam cells). Recruited T cells release interferon-γ, which inhibits SMC collagen synthesis and further fuels inflammation.
Step 5 - Advanced Plaque Formation
A fully developed atheromatous plaque has three principal components:
| Component | Contents |
|---|---|
| Fibrous cap | SMCs embedded in dense collagen + proteoglycans; covered by intact (or ulcerated) endothelium |
| Shoulder region | Cellular zone with macrophages, T cells, SMCs - most metabolically active part |
| Necrotic core | Cholesterol crystals ("clefts"), oxLDL, foam cells, necrotic debris, fibrin, calcifications |
Additional features:
- Neovascularization (plaque angiogenesis) from adventitial vasa vasorum - can bleed into the plaque (intraplaque hemorrhage)
- Calcification within older plaques
- Plaques are eccentric on cross-section (don't involve the full circumference)
Predominant sites (descending order of severity): infrarenal abdominal aorta > coronary arteries > popliteal arteries > internal carotid arteries > circle of Willis vessels.
Step 6 - Plaque Complications (Acute Plaque Change)
Most myocardial infarctions occur on plaques that were previously non-obstructive. The triggering event is acute plaque change:
Vulnerable ("Unstable") vs. Stable Plaques
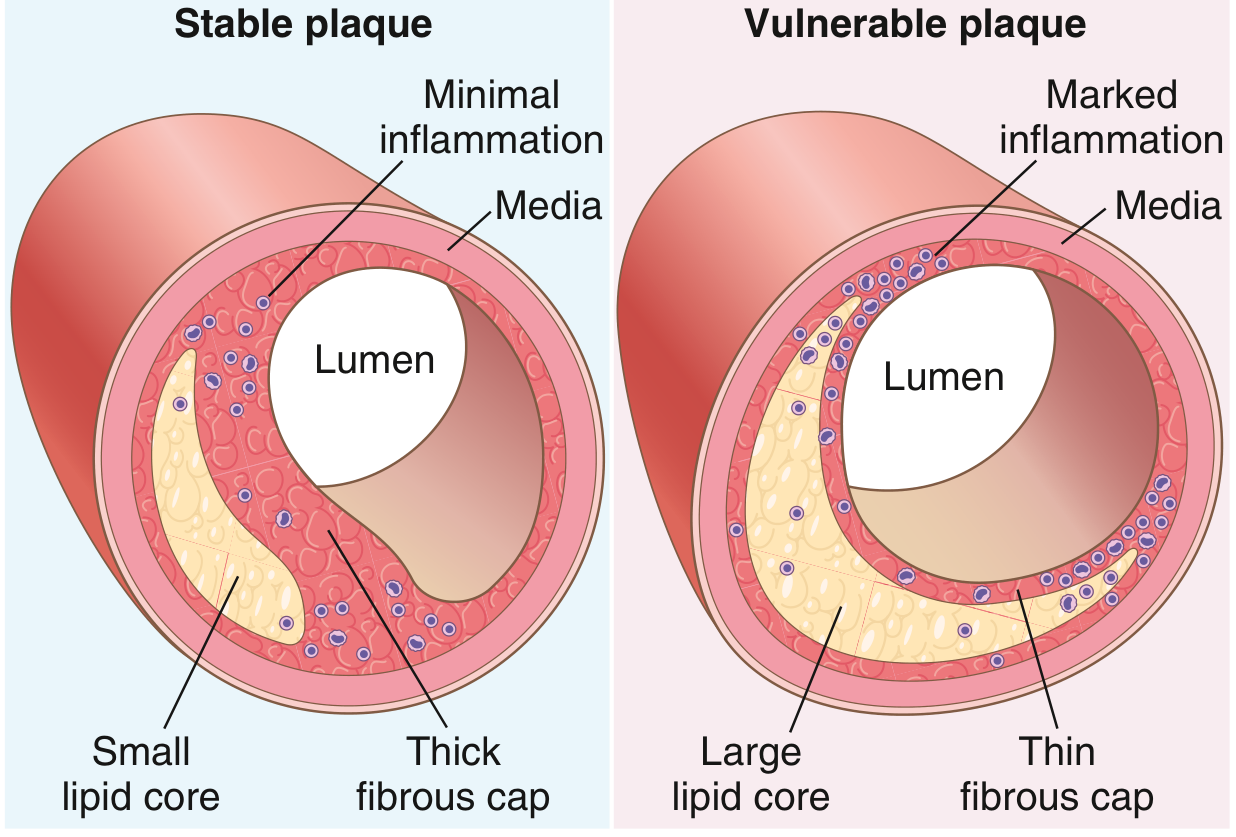
| Feature | Stable Plaque | Vulnerable Plaque |
|---|---|---|
| Fibrous cap | Thick, dense collagen | Thin |
| Lipid core | Small | Large |
| Inflammation | Minimal | Dense (macrophages, T cells) |
| Calcification | Often present | Variable |
| Risk of rupture | Low | High |
Mechanisms of Plaque Destabilization
- Macrophage-derived MMPs (matrix metalloproteinases) degrade collagen in the fibrous cap
- Interferon-γ from T cells inhibits SMC collagen synthesis - thinning the cap
- Intraplaque hemorrhage from ruptured neovessels increases lipid core volume
- Adrenergic surges (emotional stress, morning waking) increase blood pressure and shear stress on the plaque
Consequences of Plaque Rupture
- Thrombosis - exposure of the necrotic core activates platelets and the coagulation cascade; partial or total occlusion → unstable angina or MI
- Embolization - thrombus or plaque debris occludes distal vessels
- Luminal stenosis - progressive plaque growth causing chronic ischemia
- Aneurysm - media ischemia and SMC loss due to increased diffusion distance weakens the wall
Summary Diagram of Key Steps
Endothelial injury/dysfunction
↓
LDL entry → Oxidation (oxLDL)
↓
Monocyte adhesion (VCAM-1) → Macrophage
↓
Foam cell formation (SR-mediated lipid uptake)
↓
Fatty streak
↓
SMC migration & proliferation (PDGF, FGF)
+ ECM synthesis (fibrous cap)
↓
Fibrofatty atheroma
↓
Plaque progression: necrotic core enlarges,
cap thins, inflammation persists
↓
Vulnerable plaque → Rupture → Thrombosis
↓
ACS / MI / Stroke / Peripheral ischemia
Key Molecular Players
| Molecule | Role |
|---|---|
| oxLDL | Activates macrophages, cytotoxic, chemotactic |
| VCAM-1 / ICAM-1 | Monocyte/T cell adhesion to dysfunctional EC |
| MCP-1 (CCL2) | Monocyte chemoattractant |
| Scavenger receptors (SR-A, CD36) | Unregulated foam cell formation |
| PDGF, FGF, TGF-β | SMC migration and proliferation |
| MMPs | Fibrous cap degradation (plaque instability) |
| IFN-γ | Inhibits SMC collagen synthesis |
| NO | Atheroprotective: vasodilation, anti-adhesion, anti-platelet |
| HDL | Reverse cholesterol transport, antioxidant |
Sources:
- Robbins & Kumar Basic Pathology - Chapter 8, pp. 314-320 (Response to Injury Hypothesis, Plaque Morphology, Acute Plaque Change)
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 11, pp. 473-475 (Atherosclerosis summary)
- Fuster and Hurst's The Heart, 15th Edition - Chapter 16 (Endothelial Dysfunction, Smoking and Atherogenesis)
Recent evidence (2025) highlights the NLRP3 inflammasome as a key therapeutic target in atherosclerosis - phytochemicals that inhibit NLRP3 can reduce plaque inflammation (PMID 40443656). A 2025 systematic review also updates the role of oxLDL-driven inflammatory cell activation as a central mechanism (PMID 41193878).
This is a shared conversation. Sign in to Orris to start your own chat.